| Posted: May 06, 2016 |
More accurate predictions for harvesting hydrogen with iridium oxide nanoparticles
(Nanowerk News) Iridium oxide (IrO2) nanoparticles are useful electrocatalysts for splitting water into oxygen and hydrogen — a clean source of hydrogen for fuel and power. However, its high cost demands that researchers find the most efficient structure for IrO2 nanoparticles for hydrogen production.
|
|
A study conducted by a team of researchers at the U.S. Department of Energy's (DOE's) Argonne National Laboratory, published in Journal of Materials Chemistry A ("Towards accurate prediction of catalytic activity in IrO2 nanoclusters via first principles-based variable charge force field"), describes a new empirical interatomic potential that models the IrO2 properties important to catalytic activity at scales relevant to technology development. Also known as a force field, the interatomic potential is a set of values describing the relationship between structure and energy in a system based on its configuration in space. The team developed their new force field based on the MS-Q force field.
|
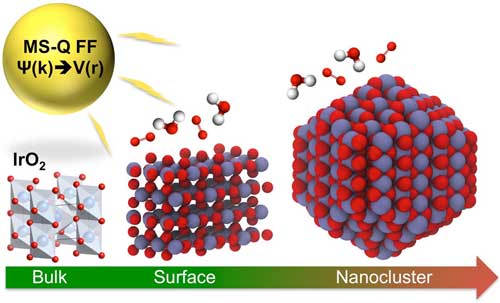 |
| Researchers from Argonne National Laboratory developed a first-principles-based, variable-charge force field that has shown to accurately predict bulk and nanoscale structural and thermodynamic properties of IrO2. Catalytic properties pertaining to the oxygen reduction reaction, which drives water-splitting for the production of hydrogen fuel, were found to depend on the coordination and charge transfer at the IrO2 nanocluster surface. (Image courtesy of Maria Chan, Argonne National Laboratory)
|
|
"Before, it was not possible to optimize the shape and size of a particle, but this tool enables us to do this," said Maria Chan, assistant scientist at Argonne's Center for Nanoscale Materials (CNM), a DOE Office of Science User Facility.
|
|
Until now, researchers have relied on the quantum mechanical modeling method based on density functional teory (DFT) to investigate the electronic structure of IrO2 on the atomic scale. However, DFT is a computationally intensive method, and while it enables some calculations of electronic behavior, DFT simulations are only capable of handling a small number of atoms. DFT models cannot scale to sizes relevant to nanotechnology components, typically 1 to 5 nanometers, at which structural and long-range behavior can influence performance.
|
|
To create a computational framework that can help engineer IrO2 structures for powerful and efficient material technologies, researchers first needed to simulate a system of realistic size. In the past, computational and materials scientists have turned to the empirical force field over DFT for larger structures. By modeling with a typical empirical force field, researchers can readily calculate a larger system but without the electronic properties modeled in DFT.
|
|
Yet, modeling IrO2 at both an applicable size and with its electric properties is critical to predicting catalytic activity because chemical reactions are influenced on the nanoscale by structure as well as charge distribution.
|
|
"In a lot of other catalysis problems, people have pointed out that the coordination number [atomic organization] describes atomic interaction," Chan said. "But we need to account for charge transfer as well."
|
|
Extending the capabilities of the typical force field, the Argonne team is the first to develop a first-principles-based, variable-charge force field for the Ir-O system. As such, this method is based on foundational physics laws ("first principles") derived from DFT calculations and dynamic charge distribution, which accounts for changes in electron behavior during a catalytic reaction. To validate the force field's accuracy, the team computed structural, mechanical, surface and thermodynamic properties for diverse IrO2 phases using DFT. These DFT calculations were used with a genetic algorithm that optimizes the force field parameters to DFT values. To carry out their validation tests and force field development, the team used Argonne's Laboratory Computing Resource Center and CNM's Carbon computing cluster.
|
|
Notably, the MS-Q force field they developed predicts oxygen binding energy at different sites — a key measure of the catalytic activity for the reaction that splits oxygen from water, as in electrocatalysis.
|
|
"We learned that the surface structure and atomic coordination affects catalytic efficiency drastically," said Fatih Sen, postdoctoral associate and lead author.
|
|
Specifically, their initial findings show that catalytic activity is strongest at the edges and corners of IrO2 nanocrystals due to long-range electrostatics, in addition to the local coordination of atoms (as characterized by coordination number), which has been emphasized in previous research.
|