| Apr 08, 2011 |
Are we only a hop, skip and jump away from controlled molecular motion?
|
|
(Nanowerk News) We may very well be, according to a study in this month's Nature Chemistry ("Gradient-driven motion of multivalent ligand molecules along a surface functionalized with multiple receptors"). Controlling how molecules move on surfaces could be the key to more potent drugs that block the attachment of viruses to cells, and will also speed development of new materials for electronics and energy applications. The study is the culmination of a EU-funded collaboration between Tyndall National Institute, UCC researcher Dr. Damien Thompson and colleagues at University of Twente in the Netherlands. Dr. Thompson performed computer simulations that enabled a greater understanding of how two-legged molecules move along patterned surfaces, in a kind of molecular hopscotch.
|
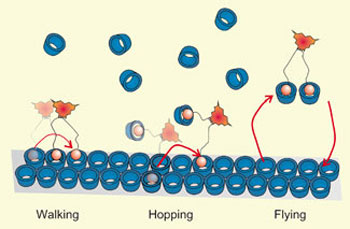 |
| The mechanism of molecular motion changes as the environment changes; in this case, due to competition from free receptors (blue) in solution (c).
|
|
Widespread industrial uptake of nanotechnology requires cheap, easy and robust solutions that allow manipulation of matter at the smallest scales and so a key enabling feature will be the ability to move material around molecule by molecule. One of the major difficulties is the very different physics that operates at the scale of atoms and molecules; water, for example, feels more like treacle to a molecule, and molecules tend to huddle and stick together due to microscopic forces between their atoms. Dr. Thompson explains: "The experiments performed by the group at Twente were very elegant. They involved making two-legged molecules and using a fluorescence microscope to watch how they move along a wet surface. The molecules are hydrophobic, meaning they don't like water, and the surface was pockmarked with hydrophobic cavities so a weak glueing interaction, based on a mutual dislike of water, drives the interaction between the molecules and the surface.
|
|
While the energetics of this type of interaction was worked out over a decade ago by George Whitesides's group at Harvard, it's usefulness for materials development was limited because little was known until now on the paths that the molecules take".
|
|
Because the molecules have multiple legs, they display a surprisingly rich behaviour at the surface, beyond simply attaching/detaching, with Dr. Thompson's computer simulations complementing the experiments and showing the different mechanisms by which the molecules move. The motion switches from walking to hopping to flying, as the environment changes.
|
|
Dr. Thompson continues: "Access to high performance computing facilities enabled us to model the different pathways and aid interpretation of the microscopy results. We ran most of the simulations on our own Science Foundation Ireland-supported computing clusters at Tyndall, and also did a few larger-scale calculations at the Irish Center for High End Computing. It's an exciting time for research as experiments and simulations are finally on the same page; the experiments can finally drill down far enough to see molecule-scale features while advances in computing mean we can routinely model systems composed of hundreds of thousands, and even millions, of atoms".
|