| Jul 13, 2017 |
Feedback from thousands of designs could transform protein engineering
|
|
(Nanowerk News) The stage is set for a new era of data-driven protein molecular engineering as advances in DNA synthesis technology merge with improvements in computational design of new proteins.
|
|
This week's Science ("Global analysis of protein folding using massively parallel design, synthesis, and testing") reports the largest-scale testing of folding stability for computationally designed proteins, made possible by a new high-throughput approach.
|
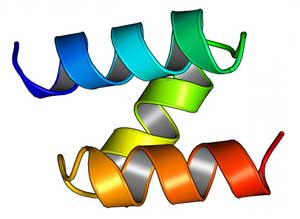 |
| A model of a computationally designed mini-protein from a large-scale study to test structural stability. (Image: UW Medicine Institute for Protein Design)
|
|
The scientists are from the UW Medicine Institute for Protein Design at the University of Washington in Seattle and the University of Toronto in Ontario.
|
|
The lead author of the paper is Gabriel Rocklin, a postdoctoral fellow in biochemistry at the University of Washington School of Medicine. The senior authors are Cheryl Arrowsmith, of the Princess Margaret Cancer Center, the Structural Genomics Consortium and the Department of Medical Biophysics at the University of Toronto, and David Baker, UW professor of biochemistry and a Howard Hughes Medical Institute investigator.
|
|
Proteins are biological workhorses. Researchers want to build new molecules, not found naturally, that can perform tasks in preventing or treating disease, in industrial applications, in energy production, and in environmental cleanups.
|
|
"However, computationally designed proteins often fail to form the folded structures that they were designed to have when they are actually tested in the lab," Rocklin said.
|
|
In the latest study, the researchers tested more than 15,000 newly designed mini-proteins that do not exist in nature to see whether they form folded structures. Even major protein design studies in the past few years have generally examined only 50 to 100 designs.
|
|
"We learned a huge amount at this new scale, but the taste has given us an even larger appetite," said Rocklin. "We're eager to test hundreds of thousands of designs in the next few years."
|
|
The most recent testing led to the design of 2,788 stable protein structures and could have many bioengineering and synthetic biology applications. Their small size may be advantageous for treating diseases when the drug needs to reach the inside of a cell.
|
|
Proteins are made of amino acid chains with specific sequences, and natural protein sequences are encoded in cellular DNA. These chains fold into 3-dimensional conformations. The sequence of the amino acids in the chain guide where it will bend and twist, and how parts will interact to hold the structure together.
|
|
For decades, researchers have studied these interactions by examining the structures of naturally occurring proteins. However, natural protein structures are typically large and complex, with thousands of interactions that collectively hold the protein in its folded shape. Measuring the contribution of each interaction becomes very difficult.
|
|
The scientists addressed this problem by computationally designing their own, much simpler proteins. These simpler proteins made it easier to analyze the different types of interactions that hold all proteins in their folded structures.
|
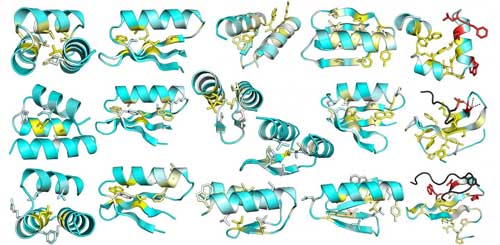 |
| This image is from a comprehensive mutational analysis of stability in designed and natural proteins. The average change in stability due to mutating each position in 13 designed proteins is depicted on the design model structures. Yellow indicates positions where mutations are most destabilizing; positions where there is little effect are blue. (Image: UW Medicine Institute for Protein Design) (click on image to enlarge)
|
|
"Still, even simple proteins are so complicated that it was important to study thousands of them to learn why they fold," Rocklin said. "This had been impossible until recently, due to the cost of DNA. Each designed protein requires its own customized piece of DNA so that it can be made inside a cell. This has limited previous studies to testing only tens of designs."
|
|
To encode their designs of short proteins in this project, the researchers used what is called DNA oligo library synthesis technology. It was originally developed for other laboratory protocols, such as large gene assembly. One of the companies that provided their DNA is CustomArray in Bothell, Wash. They also used DNA libraries made by Agilent in Santa Clara, Calif., and Twist Bioscience in San Francisco.
|
|
By repeating the cycle of computation and experimental testing over several iterations, the researchers learned from their design failures and progressively improved their modeling. Their design success rate rose from 6 percent to 47 percent. They also produced stable proteins in shapes where all of their first designs failed.
|
|
Their large set of stable and unstable mini-proteins enabled them to quantitatively analyze which protein features correlated with folding. They also compared the stability of their designed proteins to similarly sized, naturally occurring proteins.
|
|
The most stable natural protein the researchers identified was a much-studied protein from the bacteria Bacillus stearothermophilus. This organism basks in high temperatures, like those in hot springs and ocean thermal vents. Most proteins lose their folded structures under such high temperature conditions. Organisms that thrive there have evolved highly stable proteins that stay folded even when hot.
|
|
"A total of 774 designed proteins had a higher stability scores than this most protease-resistant monomeric protein," the researchers noted. Proteases are enzymes that break down proteins, and were essential tools the researchers used to measure stability for their thousands of proteins.
|
|
The researchers predict that, as DNA synthesis technology continues to improve, high-throughput protein design will become possible for larger, more complex protein structures.
|
|
"We are moving away from the old style of protein design, which was a mix of computer modeling, human intuition, and small bits of evidence about what worked before." Rocklin said. "Protein designers were like master craftsmen who used their experience to hand-sculpt each piece in their workshop. Sometimes things worked, but when they failed it was hard to say why. Our new approach lets us collect an enormous amount of data on what makes proteins stable. This data can now drive the design process."
|
