| Posted: Apr 22, 2014 | |
A novel approach to Density Functional Theory |
|
| (Nanowerk Spotlight) In the past decades, the Density Functional Theory (DFT) has been very successful in helping chemists and physicists understand the properties of matter at extremely small scales. Despite this theory being based on some rather strong assumptions – such as, for instance, local density approximations for exchange-correlation effects – it represents an important theoretical tool which is now used on a daily basis. | |
| For example, researchers have demonstrated through DFT calculations: | |
|
|
|
| The success of DFT relies on the fact that the challenging quantum N-body problem is reduced to a set of N single-body Schrödinger equations which are numerically easier to treat, where the N equations are coupled by a density functional which models the exchange-correlation effects. | |
| Now, even assuming that we have an exact functional for these effects, some problems still remain in the standard implementation of DFT: 1) the theory is based on the Schrödinger formalism, i.e. complex valued wave-functions which have no counterpart in the real world; and 2) it is difficult to scale these deterministic algorithms (usually based on finite difference and/or finite element methods), i.e. complex quantum systems consisting of a relatively big number of atoms is difficult to simulate even on parallel machines. | |
| Very recently, exciting results have been achieved by two scientists, Dr. J.M. Sellier and Prof. I. Dimov, at the Institute of Information and Communication Technologies (IICT) of the Bulgarian Academy of Sciences in Sofia. Reporting their findings in Journal of Computational Physics ("A Wigner Monte Carlo approach to density functional theory"), they have been able to use the Wigner formalism of Quantum Mechanics to simulate chemical systems. | |
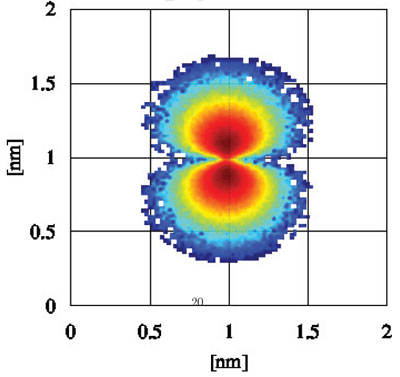 |
|
| Density of an electron in Boron 2p orbital calculated by the Wigner Monte Carlo DFT method (logarithmic scale). (Image: Dr. Sellier) | |
| This represents an important result since the researchers' approach is based on Monte Carlo techniques – a mathematical method which strongly relies on the generation of independent random numbers to solve very complicated integro-differential equations. | |
| "In our work, we propose a variant of the standard DFT, where the set of coupled single-electron Schrödinger equations in the Kohn-Sham system is substituted by an equivalent set of single-electron Wigner equations, which are solved by the Wigner Monte Carlo method," Sellier tells Nanowerk. "There are two big advantages using this approach: 1) the theory is based on the Wigner formalism, i.e. on intuitive (quasi) distribution functions which are easy to understand when compared to experimental results; 2) it scales incredibly well being based on Monte Carlo techniques, i.e. it paves the way towards the simulation of very complex chemical and physical systems at a quantum level." | |
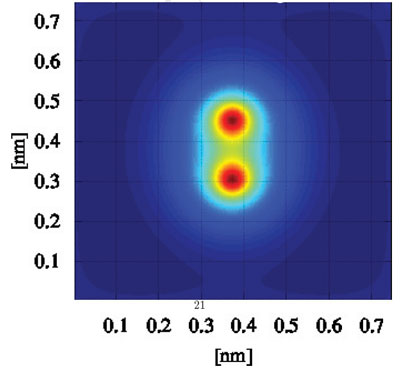 |
|
| This image shows the probability density in a Hydrogen (H2) molecule. In particular it is possible to see how the method predicts correctly the creation of a chemical bond between the two hydrogen atoms. (Image: Dr. Sellier) | |
| This represents an important result since the researchers' approach is based on Monte Carlo techniques – a mathematical method which strongly relies on the generation of independent random numbers to solve very complicated integro-differential equations. | |
| Sellier and Dimov validate this new approach by applying it to the study of three different quantum systems: a lithium atom; a Boron atom; and an hydrogenic molecule in two different configurations (close and far apart nuclei). Furthermore, in the last system, a comparison with the standard DFT is performed. | |
| If you are interested in digging deeper into this subject, you can find more details along with a list of publications and animations at www.nano-archimedes.com. | |
| Dr. Sellier and Prof. Dimov are very enthusiastic about this new approach, which is being perceived as an important evolution of DFT by the scientific community. It paves the way towards scalable simulations of complex chemical and nanotechnology systems that can now run on relatively small parallel machines. | |
 By
Michael
Berger
– Michael is author of three books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Technology,
Nanotechnology: The Future is Tiny, and
Nanoengineering: The Skills and Tools Making Technology Invisible
Copyright ©
Nanowerk LLC
By
Michael
Berger
– Michael is author of three books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Technology,
Nanotechnology: The Future is Tiny, and
Nanoengineering: The Skills and Tools Making Technology Invisible
Copyright ©
Nanowerk LLC
|
|
|
Become a Spotlight guest author! Join our large and growing group of guest contributors. Have you just published a scientific paper or have other exciting developments to share with the nanotechnology community? Here is how to publish on nanowerk.com. |
|