| Posted: Dec 07, 2016 |
Machine learning enables predictive modeling of 2-D materials
(Nanowerk News) Machine learning, a field focused on training computers to recognize patterns in data and make new predictions, is helping doctors more accurately diagnose diseases and stock analysts forecast the rise and fall of financial markets. And now materials scientists have pioneered another important application for machine learning -- helping to accelerate the discovery and development of new materials.
|
|
Researchers at the Center for Nanoscale Materials and the Advanced Photon Source, both U.S. Department of Energy (DOE) Office of Science User Facilities at DOE's Argonne National Laboratory, announced the use of machine learning tools to accurately predict the physical, chemical and mechanical properties of nanomaterials.
|
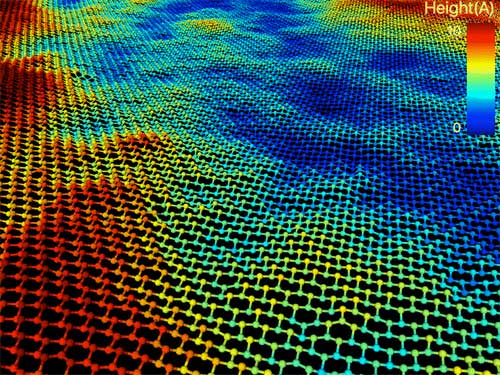 |
| The badlands of stanene: Stanene is softer and consequently much more rippled than its cousins graphene and silicene. (Image by Mathew Cherukara, Badri Narayanan and Subramanian Sankaranarayanan/Argonne National Laboratory)
|
|
In a study published in The Journal of Physical Chemistry Letters ("Ab Initio-Based Bond Order Potential to Investigate Low Thermal Conductivity of Stanene Nanostructures"), a team of researchers led by Argonne computational scientist Subramanian Sankaranarayanan described their use of machine learning tools to create the first atomic-level model that accurately predicts the thermal properties of stanene, a two-dimensional (2-D) material made up of a one-atom-thick sheet of tin.
|
|
The study reveals for the first time an approach to materials modeling that applies machine learning and is more accurate at predicting material properties compared to past models.
|
|
"Predictive modeling is particularly important for newly discovered materials, to learn what they're good for, how they respond to different stimuli and also how to effectively grow the material for commercial applications -- all before you invest in costly manufacturing," said Argonne postdoctoral researcher Mathew Cherukara, one of the lead authors of the study.
|
|
Traditionally, atomic-scale materials models have taken years to develop, and researchers have had to rely largely on their own intuition to identify the parameters on which a model would be built. But by using a machine learning approach, Cherukara and fellow researchers were able to reduce the need for human input while shortening the time to craft an accurate model down to a few months.
|
|
"We input data obtained from experimental or expensive theory-based calculations, and then ask the machine, 'Can you give me a model that describes all of these properties?'" said Badri Narayanan, an Argonne postdoctoral researcher and another lead author of the study. "We can also ask questions like, 'Can we optimize the structure, induce defects or tailor the material to get specific desired properties?'"
|
|
Unlike most past models, the machine learning model can capture bond formation and breaking events accurately; this not only yields more reliable predictions of material properties (e.g. thermal conductivity), but also enables researchers to capture chemical reactions accurately and better understand how specific materials can be synthesized.
|
|
Another advantage of building models using machine learning is the process is not material-dependent, meaning researchers can look at many different classes of materials and apply machine learning to various other elements and their combinations.
|
|
The computational model Cherukara, Narayanan and their colleagues have developed describes stanene, a structure made of tin that has caught the eye of researchers in recent years. Interest in stanene mirrors a growing interest in 2-D materials evolving from the 2004 discovery of graphene, a single-layer arrangement of carbon with attractive electronic, thermal and mechanical properties. While stanene remains far from commercialization, researchers find it promising for applications in thermal management (the regulation of heat) across some nanoscale devices.
|

