| Jul 30, 2018 |
Supercomputing the 'how' of chemical reactions(Nanowerk News) Sometimes, when experimental scientists get their hands on a supercomputer, it can change the course of their careers and open up new questions for exploration. |
| This was the case with Abdurrahman and Tülay Atesin, husband and wife chemists, collaborators and professors at the University of Texas Rio Grande Valley. Experimentalists by training, when they moved to Texas in 2013, a colleague told them that through the University of Texas Research Cyberinfrastructure initiative they had free access to some of the advanced computing systems in the world at the Texas Advanced Computing Center (TACC). |
| "We weren't planning on doing intensive computational studies, but once we were introduced to resources at TACC it opened our research horizons to collaborate with other groups within UT system and other parts of the country," said Tülay. "It has been extremely helpful for both of our research groups and our research productivity. Having TACC resources helped us a great amount in continuing our research." |
| Over the past five years, the Atesins have used TACC supercomputers - initially Longhorn, Lonestar and Stampede, then Lonestar5 and now Stampede2 - to study organometallic compounds: chemical compounds that contain bonds between a carbon atom of an organic molecule and a metal. |
| Organometallic compounds are widely used in industrial applications and serve as catalysts for the production of polymers, pharmaceuticals, and many other types of practical products. However, it isn't the end products that interest the Atesins as much as the process molecules go through to get there. |
| Their most recent research deals with the element, palladium, and its role in synthesizing cyclopentenones - five-membered rings which play a role in diverse compounds like the scent of jasmine and prostaglandins, a lipid that has hormone-like effects in animals. |
 |
| Animation showing the lowest energy transition state for the formation of cyclopentenones. |
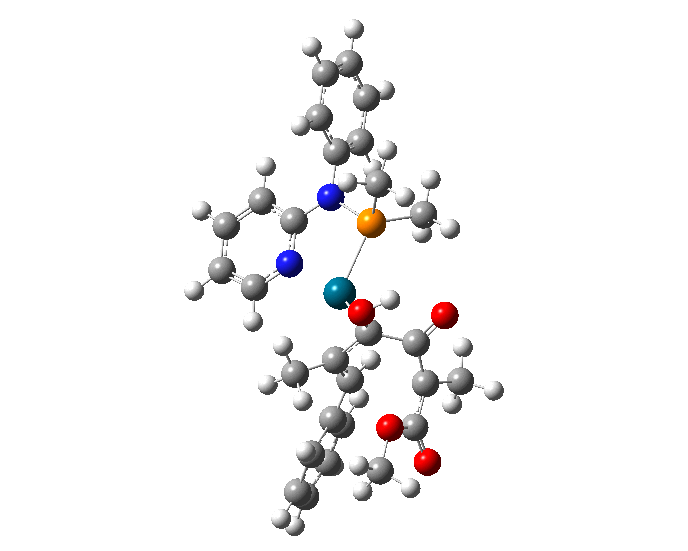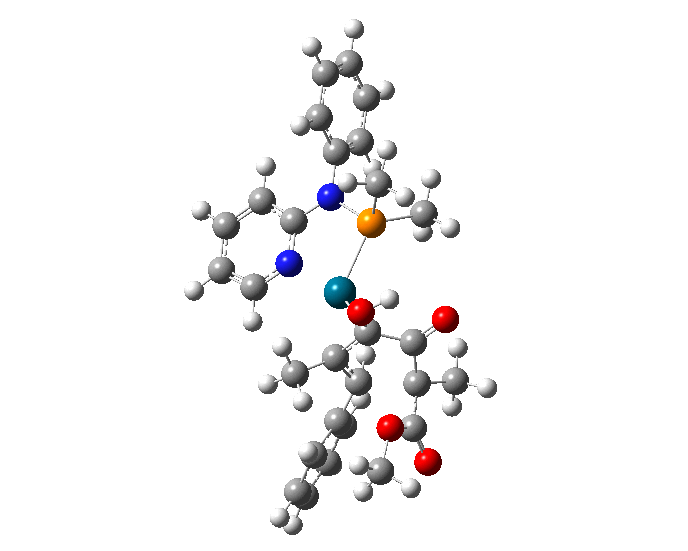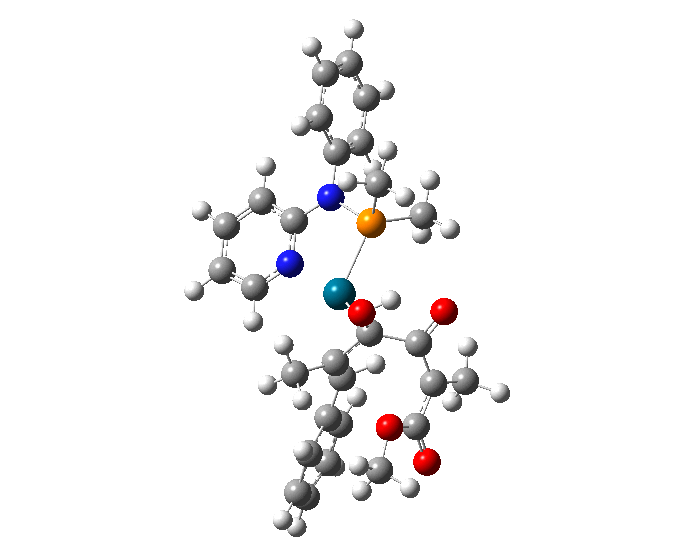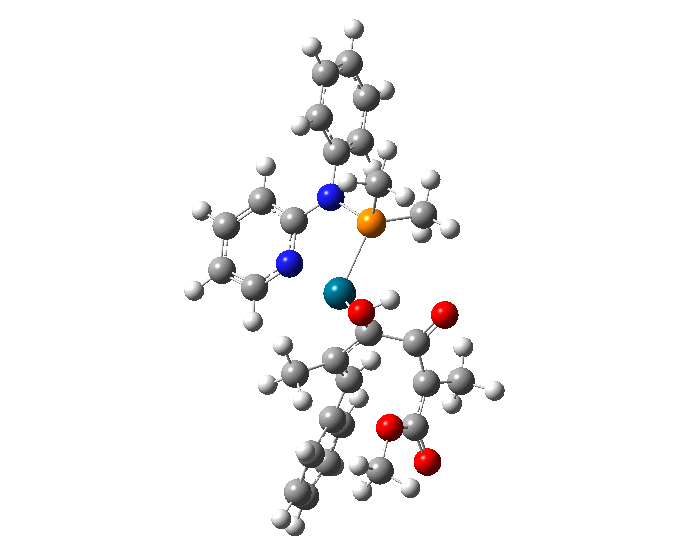
| In July 2018, the Atesins, working with UTRGV collaborators Oscar Rodriguez, Diego Rivera, and Lohany Garcia, published the results of a study in Computational and Theoretical Chemistry ("Conformational analysis of a TADDOL-based phosphoramidite P,N ligand in a palladium(II) η3-π-allyl complex") exploring the structure of a palladium catalyst in order to understand the exceptional selectivity observed in palladium-catalyzed reactions. |
| The results supported their hypothesis that the most stable form of the molecule is chair-shaped and that repulsion between this conformation and the substrate (the substance on which the molecule acts) dictates which ultimate final product forms. |
| To arrive at this conclusion, the researchers performed molecular mechanical calculations to generate 53 unique structures that could potentially represent phosphoramidites -- a class of versatile molecules with a range of applications for catalysis. They then used quantum mechanical calculations on the Stampede supercomputer at TACC to further analyze these structures and determine which had the lowest energy (and therefore were the most likely to occur in nature) and to assess the forces at work when they reacted. |
| The research findings can be used to understand the observed selectivity in many impactful palladium-catalyzed reactions and to guide the synthesis of new and improved variants of this important catalyst family. |
| In separate research reported in Organometallics in September 2017 ("It Is Not Just Up to the Substrate: Palladium(0) Cyclizes Nazarov Substrates through Intramolecular Allylic Alkylation"), they explained the mechanism of a reaction that many thought was a "Nazarov" reaction since the reactants and the products of the reaction are the same as a classical "Nazarov" reaction. |
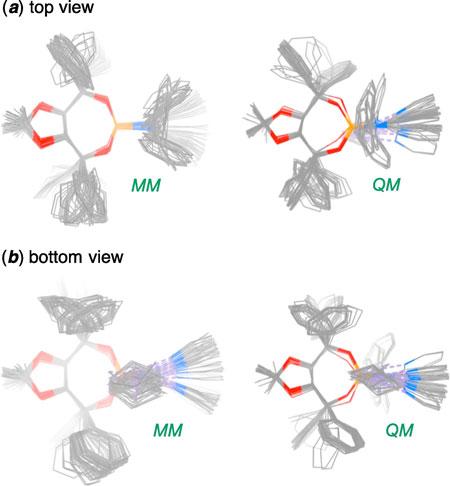 |
| A comparison of the overlays of the wire representations of all of the 53 molecular conformations derived through molecular mechanical and quantum mechanical calculations. (a) top view and (b) bottom view. |
| "Everyone in the field thought that palladium(0) does not function as a Lewis acid, but its role was not clear," Tülay Atesin said. In 2012, when the reaction was first reported, "the mechanism was unknown. So, we studied what the mechanism could be." |
| What they discovered was the first known example of the use of an "asymmetric allylic alkylation reaction" for the synthesis of a chiral cyclopentenone. (Chirality is a characteristic of a molecule that means it cannot be superimposed on its mirror image.) |
| To uncover the mechanism, they used a computational method known as density functional theory, or DFT, according to Abdurrahman. |
| "With DFT, we input a beginning structure and a final structure that we've determined experimentally, and we try different routes and approaches to see how you can connect those," he said. "This requires some chemical intuition into what the metal can do and some luck as well." |
| DFT simulations on Stampede revealed the proton transfer and ring formation processes, as well as the energy levels and geometry changes of the constituent molecules. They also performed simulations with and without palladium - essentially running blank experiments that are impossible to perform in the lab. The researchers then visualized these simulations to understand what was happening to the molecules at all of the intermediate stages. |
| "It's difficult to isolate reaction intermediates and transition states in the lab, because they're so short-lived," Tülay said. However, computer simulations can show each potential step of the process, including intermediates, which helps scientists generate new hypotheses and theories about how the reaction occurs. |
| "We never thought we'd have discovered these intermediates," Tülay said. "We weren't looking for an allylic alkylation reaction. We were asking, 'What if the metal is here? What if it's there?' And that led us to see what other possibilities were out there in terms of the mechanisms." |
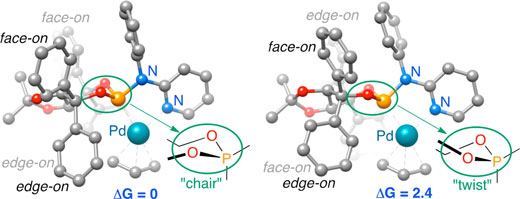 |
| Researchers used molecular mechanical and quantum mechanical calculations to determine the most stable conformations of a TADDOL-based phosphor-amidite P,N ligand coordinated to a palladium(II) η3-π-allyl fragment. The results of this research can be used to guide the synthesis of new and improved variants of this important catalyst family. |
| The most important advantage of the process they unearthed is that it is 100 percent efficient and forms a complex without the addition of other substances. Research in this vein may one day allow chemists to synthesize materials - in particular natural compounds and other bioactive molecules with all-carbon-atom centers -- that are currently difficult to create. It may even lead to entirely new types of chemical reactions that are not currently known or used. |
| Mechanistic studies using TACC resources give the Atesins a competitive edge in their work, Tülay said. "It takes our research to a higher-level than just working on experimental research. It also impacts how we design our next set of experiments." |
| Source: University of Texas at Austin, Texas Advanced Computing Center |
|
Subscribe to a free copy of one of our daily Nanowerk Newsletter Email Digests with a compilation of all of the day's news. |

