| Posted: Sep 12, 2018 |
Predicting diffusion data(Nanowerk News) The diffusion of light elements into metals has been efficiently modeled by A*STAR researchers using a machine learning approach (Computational Materials Science, "Prediction of interstitial diffusion activation energies of nitrogen, oxygen, boron and carbon in bcc, fcc, and hcp metals using machine learning"). |
| Solid-state diffusion, in which atoms migrate through the lattice of a host material, underpins a variety of important processes that range from undesirable (corrosion) to useful (metal-joining processes). In one mechanism called ‘interstitial diffusion’, light elements, such as nitrogen, move through lattices made up of much bigger atoms, such as metals, by squeezing between them. Yingzhi Zeng and colleagues at the A*STAR Institute of High Performance Computing have now developed a rapid predictive model for this phenomenon. |
 |
| Flowchart diagram of the prediction of diffusion data (activation energies) using the model developed through machine learning. (Image: A*STAR Institute of High Performance Computing) |
| “Typical examples of interstitial diffusion include surface hardening of steel through carburization or nitridation, and the diffusion of oxygen in titanium for the design of implant and aerospace alloys” Zeng says. This process is important to understand, but particularly difficult to probe experimentally. The challenge stems from the heavy-duty specialized equipment that is often required, and because as Zeng explains, “most experimental techniques rely on surface measurements, and so are inherently limited to a few nanometers under the surface.” |
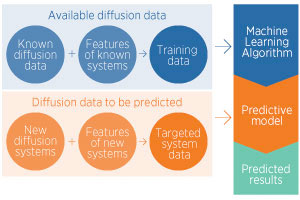
| Computational studies can circumvent these technical difficulties; first-principle methods have been shown to reliably predict diffusion transport rates, but they are time-consuming. Yingzhi Zeng and co-workers have dramatically sped up calculations of diffusion activation energies — the energy required for a light element to move through its host lattice — through machine learning. |
| They first ‘trained’ a model on a set of existing data, consisting of experimental activation energies complemented with first-principle calculations. The dataset was selected for consistency: for example only high temperatures and small solute concentrations were considered. 94 systems were used, each consisting of one solute (boron, carbon, oxygen or nitrogen) diffusing through a metal host adopting one of three most widespread lattice arrangements: body center cubic (bcc), face-centered cubic (fcc) or hexagonal close packed (hcp). |
| The accuracy of the model was verified by using it to predict known activation energies, and comparing the calculated results with the experimental values. It was then used to calculate activation energies for systems for which no experimental data have been reported. “Our predicted results have offered large amounts of reliable data — 554 new sets of diffusion data covering almost all the metals in the periodic table with the three common crystal structures of bcc, fcc, and hcp — for the conditions that are most commonly used in experiments”, Zeng says. |
| The immediate aim of the study is two-fold: to go on to predict transport rates in materials, and to gain insight into the factors driving the diffusion process. But the team won’t stop there. “We are planning to develop a mobility database for materials microstructure simulation”, Zeng says. |
| Source: A*STAR |
|
Subscribe to a free copy of one of our daily Nanowerk Newsletter Email Digests with a compilation of all of the day's news. |

