| Sep 25, 2023 |
Towards computational design of molecules with desired properties
(Nanowerk News) Prof. Alexandre Tkatchenko and his research team at the University of Luxembourg in collaboration with Cornell University and the Argonne National Laboratory have introduced a novel “freedom of design” principle in the chemical compound space (CCS) – the unfathomably vast space populated by all possible atomic compositions and their geometries. The team then showed that this principle has important implications for enabling the rational design of molecules with a desired set of properties.
|
|
The exploration of the remarkably vast space of molecules and materials with data-driven approaches has inspired countless academic and industrial initiatives to seek out the fundamental relationships that exist between the structural signatures of molecules and their physicochemical properties. While there has been significant progress in this area, a comprehensive understanding of these complex relationships – even in the more manageable sector of CCS spanned by small molecules – was still lacking despite the critical importance and high relevance of such molecules throughout the chemical and pharmaceutical sciences.
|
|
“Unravelling complex relationships between molecular structures and properties would not only provide us with the tools needed to explore and characterise the molecular space, but it would also greatly advance our ability to rationally design molecules with targeted array of physicochemical properties”, says Alexandre Tkatchenko, professor of Theoretical Chemical Physics in the Department of Physics and Materials Science at the University of Luxembourg.
|
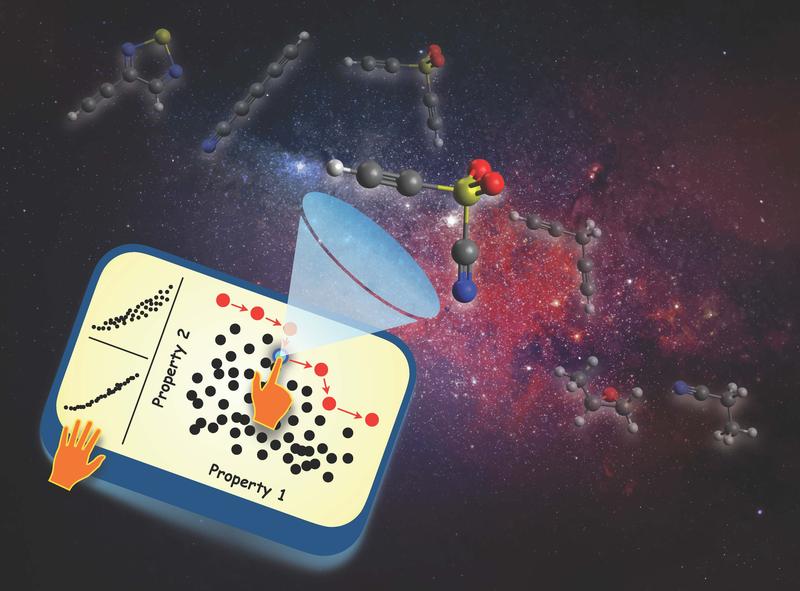 |
| Graphical depiction of the rational molecular design process, which involves a “needle-in-a-haystack” search for molecules with a desired set of properties. (Image: Leonardo Medrano Sandonas, University of Luxembourg, background image by rawpixel.com on Freepik)
|
Weak correlations enable “freedom of design”
|
|
One of the key findings of the paper, published in the journal Chemical Science ("Freedom of Design” in Chemical Compound Space: Towards Rational in Silico Design of Molecules with Targeted Quantum-Mechanical Properties"), is that most of quantum-mechanical properties of small molecules are only weakly correlated.
|
|
“While one might initially view this finding as a challenge for rational molecular design, we argue that our analysis highlights an intrinsic flexibility – or “freedom of design” – that exists in CCS, wherein there seems to be very few limitations preventing a molecule from simultaneously exhibiting any pair of properties or for many molecules sharing an array of properties”, says Robert DiStasio Jr., professor of Theoretical Chemistry at Cornell University.
|
Searching for optimal pathways in chemical space
|
|
To explore how this intrinsic flexibility will manifest in the molecular design process, which often involves the simultaneous optimization of multiple physicochemical properties, the authors used Pareto multi-property optimisation to search for molecules with simultaneously large molecular polarisability and electronic gap, a design task of relevance for identifying novel molecules for polymeric batteries. The authors found paths through chemical space consisting of several unexpected molecules connected by structural and/or compositional changes, reflecting the freedom in the rational design and discovery of molecules with targeted property values.
|
|
“A potentially interesting next step would be to use these Pareto-optimal structures in conjunction with powerful machine learning approaches to build reliable multi-objective frameworks for a systematic navigation of hitherto unexplored chemical spaces”, explains Prof. Tkatchenko.
|
Implications for the molecular design paradigm
|
|
“By demonstrating that “freedom of design” is a fundamental and emergent property of CCS, our work has a number of important implications in the fields of rational molecular design and computational drug discovery. For one, we hope this work will challenge the chemical sciences community to consider how such intrinsic flexibility can be used to extend the dominant paradigm in the forward molecular design process. We also hope that this work will enable substantive progress towards solving the inverse molecular design problem, in which one seeks to find a molecule (or set of molecules) corresponding to a targeted array of properties”, explains Dr. Leonardo Medrano Sandonas, postdoctoral researcher in the Theoretical Chemical Physics group at the University of Luxembourg.
|
|
The combination of the insights gained from this work with advanced machine learning approaches could aid in the development of effective strategies for high-throughput screening of novel molecules tailored to a specific application, which is a prominent research direction in Prof. Tkatchenko’s group. The research team used the high-performance computing resources of the Argonne Leadership Computing Facility (ALCF), a DOE Office of Science user facility.
|

