| Dec 22, 2016 |
Light powers new chemistry for old enzymes |
| (Nanowerk News) Enzymes are nature's tools for catalyzing life's essential reactions. Though unrivaled in their efficiency and selectivity, enzymes only carry out a narrow range of natural reactions, limiting their usefulness in modern organic synthesis. |
| Now, Princeton researchers report a method that expands the realm of enzyme reactivity to a non-natural reaction and allows organic chemists to access the high selectivities offered by enzymes. |
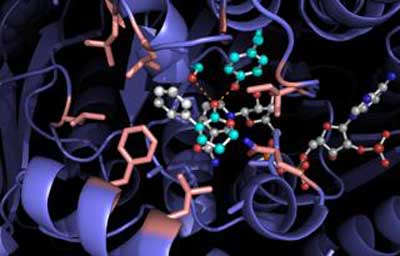 |
| Docking model for RASADH is shown. (Image: Hyster lab) |
| Published in Nature ("Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light") and led by assistant professor of chemistry Todd Hyster, the work is the first example of a reaction that employs light to activate new reactivity in an existing enzyme. The research team targeted a family of enzymes called ketoreductases that traditionally transform ketones to alcohols. Ketoreductases are dependent on a co-factor, or molecule commonly present to help drive enzyme-catalyzed reactions forward, called nicotinamide, which is known to respond to light. |
| Using a commercial kit from the company Codexis, the researchers irradiated a variety of proteins from the ketoreductase family to see if they could instead steer the chemical pathway towards a different reactivity. "These kits are a nice starting point for finding out whether your reactivity has a shot or not," Hyster said. "It really accelerated our discovery process," Hyster said. |
| Their investigations revealed that photo-excited nicotinamide-dependent ketoreductases successfully catalyzed a dehalogenation reaction, in which a halogen atom is replaced with a hydrogen atom, to produce a variety of chiral, or geometrically distinct, small molecules called lactones. |
| Through subsequent, minor mutations of the enzyme, the researchers were able to reach high reaction efficiencies and selectivities. They proposed that the good reactivity arises from a charge transfer complex and the selectivity owes to the key intermediate binding within the active site of the enzyme. |
| The reaction showed that enzymes can be coaxed to perform new chemistry and in the future Hyster's team hopes to apply their methodology to even more synthetically challenging reactions. |
| Source: Princeton University |

