| Aug 26, 2022 |
Protein structures aren't set in stone
|
|
(Nanowerk News) When you think of proteins – the enzymes, signaling molecules, and structural components in every living thing – you might think of single strands of amino acids, organized like beads on a string. But nearly all proteins consist of multiple strands folded up and bound to one another, forming complicated 3D superstructures called molecular assemblies. One of the key steps to understanding biology is discovering how a protein does its job, which requires knowledge of its structures down to the atomic level.
|
|
Over the past century, scientists have developed and deployed amazing technologies such as X-ray crystallography and cryo-electron microscopy to determine protein structure, and thereby answered countless important questions. But new work shows that understanding protein structure can sometimes be more complicated than we think.
|
|
A group of researchers from Lawrence Berkeley National Laboratory (Berkeley Lab) studying the world’s most abundant protein, an enzyme involved in photosynthesis called rubisco, showed how evolution can lead to a surprising diversity of molecular assemblies that all accomplish the same task. The findings, published in Science Advances ("Structural plasticity enables evolution and innovation of RuBisCO assemblies"), reveal the possibility that many of the proteins we thought we knew actually exist in other, unknown shapes.
|
|
Historically, if scientists solved a structure and determined that a protein was dimeric (composed of two units), for example, they might assume that similar proteins also existed in a dimeric form. But small sample size and sampling bias – unavoidable factors given that it’s very difficult to convert naturally liquid proteins into solid, crystallized forms that can be examined via X-ray crystallography – were obscuring reality.
|
|
“It's like if you walked outside and saw someone walking their dog, if you had never seen a dog before then saw a wiener dog, you’d think, ‘OK, this is what all dogs look like.’ But what you need to do is go to the dog park and see all the dog diversity that's there,” said lead author Patrick Shih, a faculty scientist in the Biosciences Area and Director of Plant Biosystems Design at the Joint BioEnergy Institute (JBEI). “One takeaway from this paper that goes beyond rubisco, to all proteins, is the question of whether or not we are seeing the true range of structures in nature, or are these biases making it seem like everything looks like a wiener dog.”
|
|
Hoping to explore all the different rubisco arrangements at the metaphorical dog park, and learn where they came from, Shih’s lab collaborated with Bioscience Area structural biology experts using Berkeley Lab’s Advanced Light Source. Together, the team studied a type of rubisco (form II) found in bacteria and a subset of photosynthetic microbes using traditional crystallography – a technique capable of atomic-level resolution – combined with another structure-solving technique, small-angle X-ray scattering (SAXS), that has lower resolution but can take snapshots of proteins in their native form when they are in liquid mixtures.
|
|
SAXS has the additional advantage of high-throughput capability, meaning that it can process dozens of individual protein assemblies in quick succession.
|
|
Previous work had shown that the better studied type of rubisco found in plants (form I) always takes an “octameric core” assembly of eight large protein units arranged with eight small units, whereas form II was believed to exist mostly as a dimer with a few rare examples of six-unit hexamers. After using these complementary techniques to examine samples of rubisco from a diverse range of microbe species, the authors observed that most form II rubisco proteins are actually hexamers, with the occasional dimer, and they discovered a never-before-seen tetrameric (four unit) assembly.
|
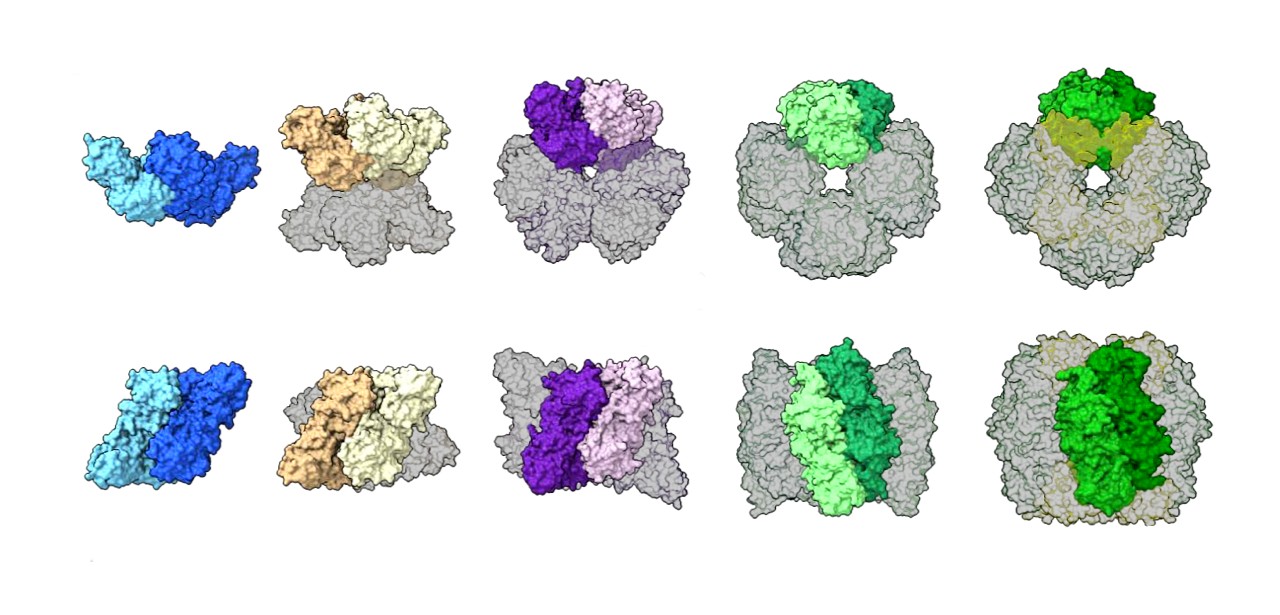 |
| A comparison of rubisco assemblies from different species, illustrating a wide range of structures including a dimer, tetramer, hexamer, octamer, and hexadecamer (16 unit protein). (Credit: Shih Lab, Berkeley Lab)
|
|
Combining this structural data with the respective protein-coding gene sequences allowed the team to perform ancestral sequence reconstruction – a computer-based molecular evolution method that can estimate what ancestral proteins looked like based on the sequence and appearance of modern proteins that evolved from them.
|
|
The reconstruction suggests that the gene for form II rubisco has changed over its evolutionary history to produce proteins with a range of structures that transform into new shapes or revert back to older structures quite easily.
|
|
In contrast, during the course of evolution, selective pressures led to a series of changes that locked form I rubisco in place – a process called structural entrenchment – which is why the octameric assembly is the only arrangement we see now. According to the authors, it was assumed that most protein assemblies were entrenched over time by selective pressure to refine their function, like we see with form I rubisco. But this research suggests that evolution can also favor flexible proteins.
|
|
“The big finding from this paper is that there’s a lot of structural plasticity,” said Shih, who is also an assistant professor at UC Berkeley. “Proteins may be much more flexible, across the field, than we’ve believed.”
|
|
After completing the ancestral sequence reconstruction, the team conducted mutational experiments to see how altering the rubisco assembly, in this case breaking a hexamer into a dimer, affected the enzyme’s activity. Unexpectedly, this induced mutation produced a form of rubisco that is better at utilizing its target molecule, CO2.
|
|
All naturally occurring rubisco frequently binds the similarly sized O2 molecule on accident, lowering the enzyme’s productivity. There is a great deal of interest in genetically modifying the rubisco in agricultural plant species to increase the protein’s affinity for CO2, in order to produce more productive and resource-efficient crops. However, there has been a lot of focus on the protein’s active site – the region of the protein where CO2 or O2 bind.
|
|
“This is an interesting insight to us because it suggests that in order to have more fruitful results engineering rubisco, we can’t just look at the simplest answer, the region of the enzyme that actually interacts with CO2,” said first author Albert Liu, a graduate student in Shih’s lab. “Maybe there are mutations outside of that active site that actually participate in this activity and can potentially change protein function in a way that we want. So that’s something that really opens doors to future avenues of research.”
|
|
Co-author Paul Adams, Associate Laboratory Director for Biosciences and Vice President for Technology at JBEI added, “The mix of techniques employed and the interdisciplinary nature of the team was a real key to success. The work highlights the power of combining genomic data and structural biology methods to study one of the most important problems in biology, and reach some unexpected conclusions.”
|

