| Oct 23, 2023 |
Researchers develop DANGER analysis tool for the safer design of gene editing
|
|
(Nanowerk News) A team of researchers has developed a software tool called DANGER (Deleterious and ANticipatable Guides Evaluated by RNA-sequencing) analysis that provides a way for the safer design of genome editing in all organisms with a transcriptome. For about a decade, researchers have used the CRISPR technology for genome editing. However, there are some challenges in the use of CRISPR.
|
|
The DANGER analysis overcomes these challenges and allows researchers to perform safer on- and off-target assessments without a reference genome. It holds the potential for applications in medicine, agriculture, and biological research.
|
Key Takeaways
|
|
DANGER analysis is a new software tool that makes genome editing safer by overcoming limitations in current CRISPR technology.
Unlike traditional methods that rely on a reference genome, DANGER works by conducting risk-averse on- and off-target assessments using RNA-sequencing data.
The tool successfully evaluated the phenotypic effects of gene editing in human cells and zebrafish brains.
DANGER is open-source and versatile, allowing for its application in various fields including medicine, agriculture, and biology.
Future plans include adapting the software for genome editing in patients and crops, aiming to establish safer editing strategies.
|
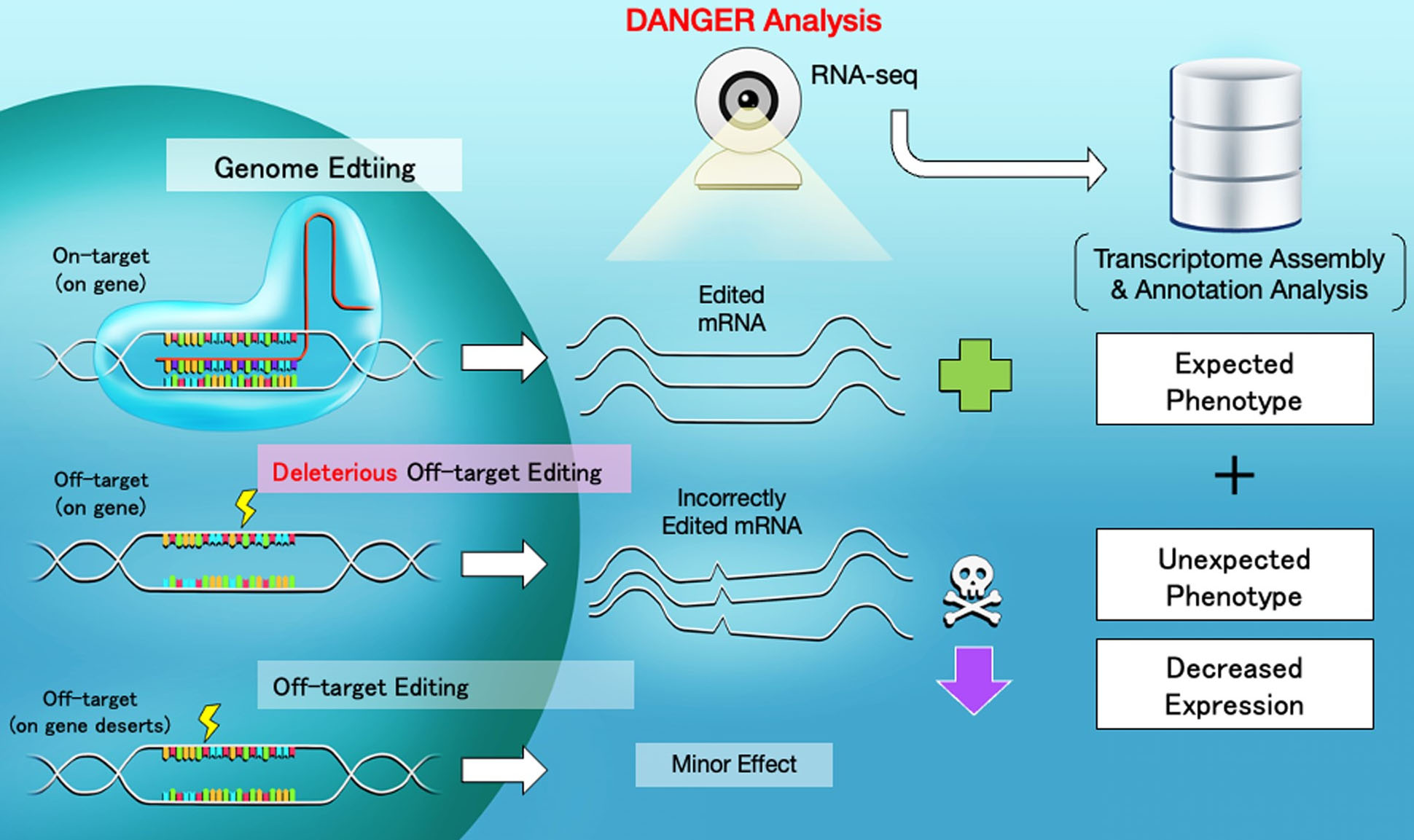 |
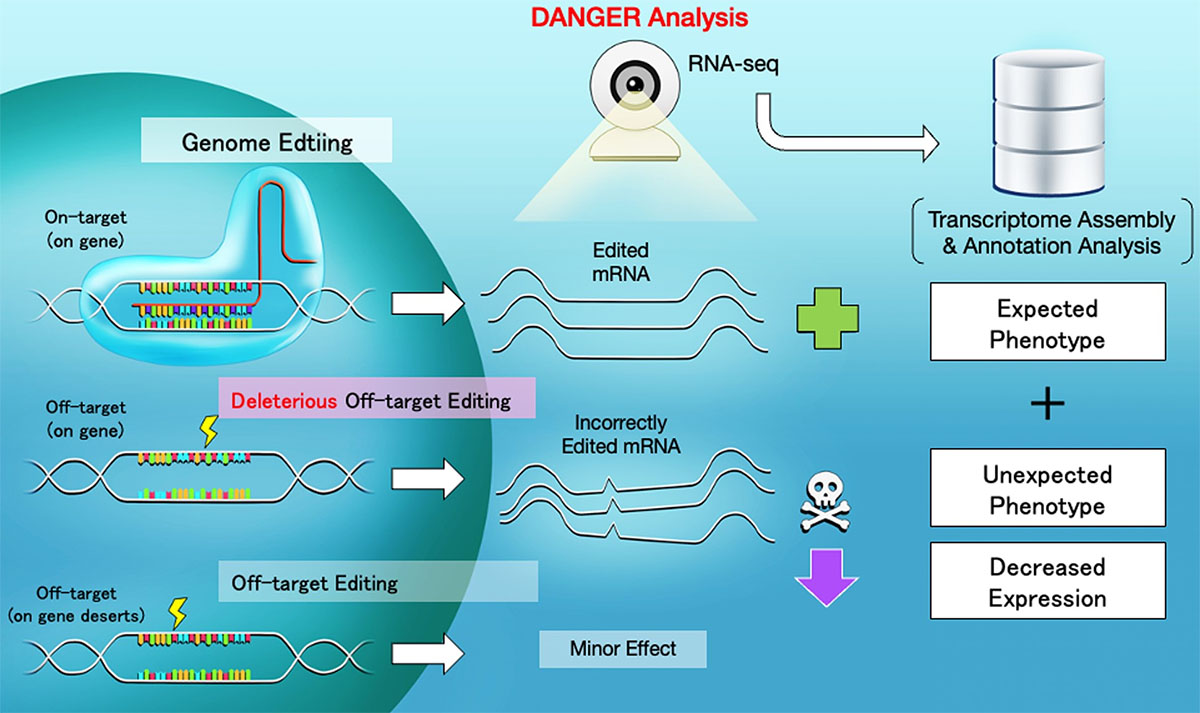
| Scheme of CRISPR-Cas9 targeting, deleterious off-target editing, and DANGER analysis. When off-target regions are affected by genome editing, unexpected changes in the mRNA quantity and sequence can emerge. The DANGER analysis is designed to analyze the effects from RNA-seq data at the Gene Ontology (GO) level. (Image: Hiroshima University) (click on image to enlarge)
|
The Research
|
|
This work is published in the journal Bioinformatics Advances ("DANGER analysis: risk-averse on/off-target assessment for CRISPR editing without a reference genome").
|
|
Genome editing, or gene editing, refers to technologies that allow researchers to change the genomic DNA of an organism. With these technologies, researchers can add, remove or alter genetic material in the genome.
|
|
CRISPR-Cas9 is a well-known gene editing technology. It has a reputation for being more accurate, faster, and less expensive than other similar technologies. However, gene editing using CRISPR technology presents some challenges. The first challenge is that the phenotypic, or observable, effects caused by unexpected CRISPR dynamics are not quantitatively monitored.
|
|
A second challenge is that the CRISPR technology generally depends on basic genomic data, including the reference genome. The reference genome is like a template that provides researchers with general information on the genome. Unexpected sequence editing with mismatches can occur. These off-target sites are always unexpected. So researchers need a way to observe factual genomic sequences and limit potential off-target effects.
|
|
“The design of genome editing requires a well-characterized genomic sequence. However, the genomic information of patients, cancers, and uncharacterized organisms is often incomplete,” said Kazuki Nakamae, an assistant professor of the PtBio Collaborative Research Laboratory at the Genome Editing Innovation Center, Hiroshima University.
|
|
The research team set out to devise a method to deal with the issues of the phenotypic effects and the dependence on a reference genome. The team’s DANGER analysis software overcomes these challenges. The team used gene-edited samples of human cells and zebrafish brains to conduct their risk-averse on- and off-target assessment in RNA-sequencing data.
|
|
The team demonstrated that the DANGER analysis pipeline achieves several goals. It detected potential DNA on- and off-target sites in the mRNA-transcribed region on the genome using RNA-sequencing data. It evaluated phenotypic effects by deleterious off-target sites based on the evidence provided by gene expression changes. It quantified the phenotypic risk at the gene ontology term level, without a reference genome. This success showed that DANGER analysis can be performed on various organisms, personal human genomes, and atypical genomes created by diseases and viruses.
|
|
The DANGER analysis pipeline identifies the genomic on- and off-target sites based on de novo transcriptome assembly using RNA-sequencing data. A transcriptome includes a collection of all the active gene readouts in a cell. With de novo transcriptome assembly, the transcriptome is assembled without the help of a reference genome. Next, the DANGER analysis identifies the deleterious off-targets. These are off-targets on the mRNA-transcribed regions that represent the downregulation of expression in edited samples compared to wild-type ones. Finally, the software quantifies the phenotypic risk using the gene ontology of the deleterious off-targets. “Our DANGER analysis is a novel software that enables quantifying phenotypic effects caused by estimated off-target. Furthermore, our tool uses de novo transcriptome assembly whose sequences can be built from RNA-sequencing data of treated samples without a reference genome,” said Hidemasa Bono, a professor at the Genome Editing Innovation Center, Hiroshima University.
|
|
Looking ahead, the team hopes to expand their research using the DANGER analysis. “We will apply the software to various genome editing samples from patients and crops to clarify the phenotypic effect and establish safer strategies for genome editing,” said Nakamae.
|
|
DANGER analysis is open-source and freely adjustable. So the algorithm of this pipeline could be repurposed for the analysis of various genome editing systems beyond the CRISPR-Cas9 system. It is also possible to enhance the specificity of DANGER analysis for CRISPR-Cas9 by incorporating CRISPR-Cas9-specific off-target scoring algorithms. The team believes that the DANGER analysis pipeline will expand the scope of genomic studies and industrial applications using genome editing.
|