ELISA (Enzyme-Linked Immunosorbent Assay): Principles, Types, and Applications
What Is ELISA?
The enzyme-linked immunosorbent assay, universally known by its acronym ELISA, is a plate-based analytical technique that detects and quantifies proteins, antibodies, hormones, and other biomolecules in biological samples by exploiting the highly specific binding between an antigen and its complementary antibody. An enzyme covalently linked to one of the binding partners converts a colorless substrate into a colored or luminescent product, generating a measurable signal that is proportional to the amount of target molecule in the sample.
ELISA was described simultaneously and independently in 1971 by Eva Engvall and Peter Perlmann at Stockholm University in Sweden and by Bauke van Weemen and Anton Schuurs in the Netherlands. Both groups were seeking a safer, less expensive alternative to radioimmunoassay, which required radioactive labels and specialized facilities. Their solution – replacing the radioactive tracer with an enzyme – preserved the sensitivity of immunoassay while eliminating the radiation hazard, and it rapidly transformed laboratory diagnosis. Within a decade, ELISA had been adopted for diagnosing viral and bacterial infections, and the format that Engvall and Perlmann named in 1971 remains one of the most widely performed assays in biomedical science.
The versatility and scalability of ELISA account for its enduring dominance. A standard 96-well microtiter plate allows 96 samples to be processed in a single experiment; 384-well formats extend this further. Automated liquid-handling systems and plate readers have made ELISA compatible with high-throughput screening. The assay requires no radioactive materials, no gel electrophoresis, and no specialized equipment beyond a microplate reader, making it accessible to virtually any biomedical laboratory. These attributes have made ELISA the reference method for an enormous range of clinical tests, from measuring hormone concentrations in blood to confirming viral infections and monitoring therapeutic proteins.
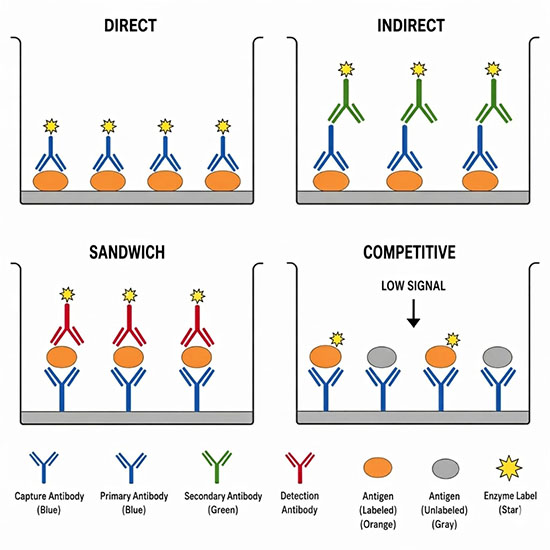
How ELISA Works
All ELISA formats share a common workflow built around three principles: immobilization of a binding partner on a solid surface, selective capture of the target molecule through antibody–antigen interaction, and enzymatic signal generation. The solid phase is almost always a 96-well polystyrene plate, which passively adsorbs proteins and antibodies through hydrophobic interactions. A blocking step – typically with bovine serum albumin or non-fat milk – saturates remaining binding sites on the plate surface and prevents non-specific attachment of assay reagents that would elevate background signal.
After blocking, the sample is added. Any target molecules present bind to the immobilized capture reagent. Unbound material is removed by a series of wash steps using phosphate-buffered saline containing a small amount of detergent. A detection antibody conjugated to a reporter enzyme is then added, and following another wash cycle, the enzyme substrate is introduced. Horseradish peroxidase (HRP), the most widely used reporter enzyme, converts the substrate TMB (3,3′,5,5′-tetramethylbenzidine) from colorless to blue; addition of a stop solution turns the product yellow and fixes the optical density. The intensity of the resulting color, measured spectrophotometrically at 450 nm, is directly proportional to the amount of target in the sample. A standard curve constructed from samples of known concentration allows unknown concentrations to be interpolated from the absorbance readings.
The choice of enzyme matters for sensitivity and dynamic range. Alkaline phosphatase, used in the original Engvall–Perlmann assay, remains an alternative to HRP and is preferred for certain quantitative applications because its product accumulates linearly over a wider concentration range. Both enzymes can also be paired with chemiluminescent or fluorescent substrates rather than colorimetric ones, extending detection sensitivity by one to two orders of magnitude compared to absorbance-based readout.
The Four Main ELISA Formats
| Format | What is coated on the plate | Best suited for | Relative sensitivity |
|---|---|---|---|
| Direct | Antigen | Screening antibody libraries; rapid detection of purified antigens | Moderate |
| Indirect | Antigen | Measuring antibody titer in serum; HIV and other serological screening | High (secondary antibody amplifies) |
| Sandwich | Capture antibody | Quantifying cytokines, hormones, and other proteins in complex samples | Highest |
| Competitive | Antigen or antibody | Small molecules and haptens that cannot be sandwiched; drug monitoring | Moderate; signal inversely proportional to concentration |
The sandwich ELISA is generally the most sensitive and specific format and dominates clinical diagnostic applications. A capture antibody coated on the plate binds one epitope of the target antigen; a second detection antibody recognizes a different epitope on the same antigen, sandwiching it between the two antibodies. Because both antibodies must bind simultaneously, cross-reactivity with structurally similar off-target molecules is substantially reduced compared to single-antibody formats. The indirect ELISA is the format most commonly used in serology, where the goal is to detect antibodies in patient serum rather than to quantify a specific protein. The competitive ELISA is preferred for small molecules – drugs, steroids, pesticide residues, and similar analytes – that present only a single epitope and therefore cannot support a sandwich configuration.
ELISA in Clinical Diagnostics
ELISA is the foundation of a large proportion of routine laboratory diagnostics. Detection of antibodies against HIV-1 and HIV-2 is performed by indirect ELISA as the primary screening step in clinical laboratories worldwide; a reactive result triggers confirmatory testing by Western blot or nucleic acid amplification. Serological diagnosis of hepatitis B and C, Lyme disease, Helicobacter pylori infection, toxoplasmosis, and many other infectious diseases relies on ELISA-based antibody detection. Sandwich ELISA formats are used for pregnancy testing (measuring human chorionic gonadotropin, hCG), thyroid function assessment (thyroid-stimulating hormone, TSH), cardiac risk profiling (troponin, B-type natriuretic peptide), and allergy evaluation (total and allergen-specific immunoglobulin E).
In oncology, ELISA quantifies circulating tumor-associated proteins that serve as biomarkers for diagnosis, staging, and treatment monitoring. Prostate-specific antigen (PSA) measurement for prostate cancer surveillance and carcinoembryonic antigen (CEA) measurement in colorectal cancer follow-up are among the highest-volume clinical ELISA tests. Autoimmune disease diagnosis also depends heavily on ELISA: detection of antinuclear antibodies, anti-double-stranded DNA antibodies in lupus, and anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis are all performed by ELISA platforms. The monoclonal antibodies used in therapeutic biologics are routinely quantified using sandwich ELISA during pharmacokinetic studies and drug monitoring.
ELISA played a central role in the COVID-19 pandemic response. Serology assays targeting the SARS-CoV-2 spike protein and nucleocapsid protein were used to characterize the antibody response in vaccinated and convalescent individuals, to assess seroprevalence across populations, and to evaluate vaccine-elicited immunity. ELISA-based antigen tests were also developed to detect viral proteins in respiratory specimens, providing a rapid and scalable complement to PCR-based nucleic acid tests during periods of high demand. The pandemic underscored ELISA’s unique combination of sensitivity, specificity, and industrial scalability.
ELISA in Research Applications
Beyond the clinical laboratory, ELISA is a core tool in basic and translational biomedical research. Cytokine profiling – measuring the concentrations of interleukins, tumor necrosis factor, interferons, and related signaling proteins in cell culture supernatants or plasma – relies almost exclusively on sandwich ELISA. Understanding which cytokines are elevated in disease states or in response to experimental treatments requires reproducible quantification across hundreds of samples, a task ELISA is uniquely suited to perform. Cell culture experiments in immunotherapy research commonly use ELISA to confirm that immune cells are secreting the expected effector molecules after activation.
Proteomics studies use ELISA in combination with mass spectrometry and other platforms for target validation: once a protein of interest is identified by discovery proteomics, ELISA provides a faster and cheaper means of measuring that protein across larger sample cohorts. Drug discovery pipelines use ELISA at multiple stages – to confirm that a candidate compound blocks a target protein’s interaction with its receptor, to measure pharmacodynamic endpoints in animal models, and to quantify drug concentrations in plasma during pharmacokinetic studies. Food safety testing uses competitive ELISA to screen for mycotoxins, pesticide residues, and allergens at concentrations as low as a few parts per billion. Environmental monitoring applies ELISA to detect bacterial toxins, herbicide residues, and veterinary drug residues in water and soil samples.
Limitations of Conventional ELISA
Despite its utility, conventional ELISA has well-recognized limitations. The dynamic range of a standard colorimetric format spans roughly two to three orders of magnitude, and samples outside this range must be diluted or concentrated before measurement. The hook effect is a particular concern in sandwich ELISA: at very high antigen concentrations, both capture and detection antibodies saturate independently rather than forming the sandwich, producing a falsely low signal. Non-specific binding is a persistent challenge with complex samples such as serum or cell lysates, and heterophile antibodies – human antibodies that bind the animal-derived reagent antibodies – are a documented source of false positives in clinical testing. The detection floor of colorimetric ELISA, approximately 1 picogram per milliliter, is too high for proteins present at trace concentrations in plasma, motivating the next-generation platforms described below.
Next-Generation and Digital ELISA
Digital ELISA overcomes the sensitivity ceiling of conventional immunoassays by detecting individual protein molecules rather than measuring ensemble signals across millions of molecules. The single-molecule array (Simoa) platform, developed around 2010, captures target proteins on paramagnetic beads, seals individual beads in femtoliter-volume wells, and counts the fraction of wells that contain an active enzyme label. Because the assay is conducted at the scale of individual molecules, it achieves sensitivity approximately 1,000-fold greater than conventional ELISA, pushing detection limits from the picogram-per-milliliter range down to femtograms per milliliter. This capability has enabled the measurement of plasma biomarkers for Alzheimer disease, multiple sclerosis, and traumatic brain injury at concentrations previously accessible only in cerebrospinal fluid.
Beyond Simoa, multiple strategies are being developed to enhance ELISA sensitivity and broaden its capabilities. Surface engineering approaches modify the microplate or bead surface to increase antibody loading density and reduce non-specific binding, improving both sensitivity and signal-to-noise ratio. Enzymatic amplification cascades – including tyramide signal amplification and rolling circle amplification – generate multiple reporter molecules per target binding event, amplifying the colorimetric or fluorescent signal. Cell-free synthetic biology is an emerging direction in which cell-free gene expression systems are coupled to immunoassay recognition elements, so that binding of the target protein triggers transcription and translation of a reporter rather than enzymatic substrate conversion; this approach can achieve attomolar sensitivity while preserving the antibody-based selectivity of ELISA.
Multiplexed ELISA platforms address a different limitation: conventional ELISA measures one analyte per well. Bead-based multiplex immunoassays, such as those using the Luminex platform, coat different populations of color-coded beads with different capture antibodies, allowing simultaneous measurement of dozens of proteins in a single sample. Proximity ligation assay (PLA) and proximity extension assay (PEA) technologies use pairs of antibodies conjugated to oligonucleotide probes that hybridize and are amplified only when both antibodies bind the same target molecule; these can be multiplexed to hundreds of proteins using next-generation sequencing or PCR readout and achieve sensitivity comparable to digital ELISA. These platforms are increasingly used in personalized medicine research and in large-scale biomarker discovery studies.
Frequently Asked Questions
What is the difference between ELISA and Western blot? Both ELISA and Western blot detect proteins using antibodies, but they serve different purposes. ELISA is a plate-based, solution-phase assay optimized for quantifying the concentration of a target protein in a large number of samples simultaneously, without separating proteins by size. Western blot separates proteins by molecular weight using gel electrophoresis before probing with antibodies, making it better suited for confirming the presence of a protein at the correct size and identifying multiple proteins in a single sample. ELISA is generally faster and more amenable to high-throughput analysis; Western blot is more confirmatory and structural.
How sensitive is ELISA compared to other detection methods? Conventional ELISA can typically detect proteins in the picogram-per-milliliter range, which is sufficient for most clinical and research applications. Next-generation digital ELISA platforms, such as single-molecule array (Simoa) technology, extend this sensitivity approximately 1,000-fold to the femtogram-per-milliliter range by counting individual immunocomplexes in femtoliter wells. Radioimmunoassay, the technology ELISA was designed to replace, achieves comparable sensitivity but requires radioactive labels and specialized handling. Modern chemiluminescent and fluorescence-enhanced ELISA formats close much of the remaining gap with standard radioimmunoassay performance.
What enzymes are used in ELISA and why? The two most widely used enzymes in ELISA are horseradish peroxidase (HRP) and alkaline phosphatase (AP). HRP is the more common choice because it is stable, small enough not to interfere with antibody binding, produces a strong colorimetric signal with substrates such as TMB, and can also be used in chemiluminescent formats that further amplify the signal. Alkaline phosphatase, the enzyme used in the original 1971 Engvall–Perlmann assay, generates a yellow color with the substrate pNPP and is preferred in some quantitative applications because it produces a more linear signal over a wider concentration range. Both enzymes are inexpensive, commercially available, and well characterized.
Can ELISA give false-positive or false-negative results? Yes. False positives can arise from non-specific binding of antibodies to the plate surface or to unrelated proteins in the sample, from cross-reactivity of the detection antibody with structurally similar targets, or from the hook effect in sandwich ELISA, where very high antigen concentrations saturate both antibodies and reduce signal. False negatives can result from sample degradation, inadequate antibody affinity, antigen denaturation during plate coating, or antigen concentrations below the assay’s detection limit. Careful optimization of blocking conditions, antibody concentrations, and wash steps – along with appropriate positive and negative controls – is essential to minimize both error types.
Can ELISA measure multiple proteins at the same time? Standard ELISA measures one analyte per well, which limits throughput when many proteins need to be measured simultaneously in the same sample. Multiplexed immunoassay platforms address this by using bead populations tagged with different fluorescent codes, each coated with a different capture antibody, allowing dozens of proteins to be quantified in a single small-volume sample. Proximity extension assay (PEA) technology can extend multiplexing to hundreds of proteins simultaneously using a sequencing readout. These multiplexed formats are widely used in biomarker discovery studies and systems biology research where a broad protein signature is more informative than a single measurement.
Further Reading
Clinical Chemistry, Enzyme Immunoassay (EIA)/Enzyme-Linked Immunosorbent Assay (ELISA)
Journal of International Medical Research, An Overview of ELISA: A Review and Update on Best Laboratory Practices for Quantifying Peptides and Proteins in Biological Fluids
