Epigenome: DNA Methylation, Histone Modifications, and Chromatin Regulation
Definition and Scope of the Epigenome
The epigenome is the complete set of chemical modifications to DNA and histone proteins that regulate gene expression across the genome without altering the underlying DNA sequence. These modifications—principally DNA methylation, histone post-translational modifications, and the positioning of nucleosomes along DNA—form a regulatory layer that determines which genes are accessible for transcription in a given cell at a given time.
Every cell type in the human body carries the same approximately 3.2 billion base pairs of DNA, yet a liver cell behaves nothing like a neuron. The epigenome explains this difference: in each cell, roughly 28 million CpG dinucleotides can be methylated and more than 100 distinct histone modifications have been catalogued, creating an immense regulatory code. During development, cells acquire distinct epigenomic signatures that silence tissue-inappropriate genes and activate cell-type-specific programs. These patterns are heritable through cell division—a core principle of epigenetics—but also responsive to environmental signals.
The NIH Roadmap Epigenomics Consortium published reference maps for 111 human epigenomes in 2015, profiling histone modification patterns, DNA methylation, DNA accessibility, and RNA expression across dozens of primary cells and tissues. This resource established the first comprehensive atlas of human epigenomic diversity and demonstrated that disease- and trait-associated genetic variants are significantly enriched in tissue-specific regulatory elements defined by epigenomic signatures.
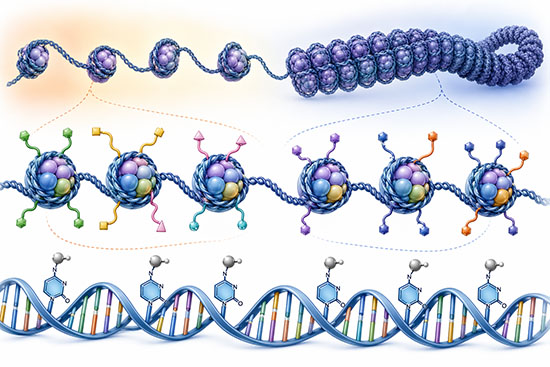
How Epigenomic Modifications Work
DNA methylation is the most extensively studied epigenomic mark. In mammals, it occurs almost exclusively at the 5-position of cytosine within CpG dinucleotides—sites where a cytosine nucleotide is followed by a guanine. The human genome contains roughly 28 million CpG sites, and their methylation status is established and maintained by a family of DNA methyltransferases (DNMTs). DNMT3A and DNMT3B catalyze de novo methylation during embryonic development, while DNMT1 copies existing methylation patterns to newly synthesized DNA strands during replication, ensuring epigenetic inheritance through cell division.
Methylation of CpG-rich regions called CpG islands, which are found near the promoters of approximately 70% of human genes, typically silences transcription by blocking the binding of transcription factors or by recruiting methyl-CpG-binding proteins that compact chromatin. Conversely, demethylation of these regions can reactivate gene expression. Active demethylation is carried out by the ten-eleven translocation (TET) family of enzymes, which oxidize 5-methylcytosine through a series of intermediates that are ultimately replaced with unmodified cytosine.
Histone modifications constitute the second major class of epigenomic marks. DNA in eukaryotic cells wraps around octamers of histone proteins—two copies each of H2A, H2B, H3, and H4—to form nucleosomes. The unstructured tails of these histones extend outward from the nucleosome core and are subject to dozens of post-translational modifications, including methylation, acetylation, phosphorylation, ubiquitination, and sumoylation. The specific combination and genomic location of these marks profoundly influence chromatin structure and gene activity.
Acetylation of histone lysine residues, catalyzed by histone acetyltransferases (HATs), generally opens chromatin and promotes transcription by neutralizing the positive charge on histone tails and weakening DNA–histone interactions. Histone deacetylases (HDACs) reverse this process and are associated with gene silencing. Histone methylation is more context-dependent: trimethylation of histone H3 at lysine 4 (H3K4me3) marks active promoters, whereas trimethylation at lysine 27 (H3K27me3), deposited by the Polycomb repressive complex 2, marks silenced developmental genes.
In embryonic stem cells, a subset of developmental gene promoters carry both H3K4me3 and H3K27me3 simultaneously—a bivalent state that keeps these genes poised for rapid activation or stable repression upon differentiation. As cells commit to specific lineages, bivalent chromatin resolves: one mark is typically lost while the other is retained, locking in the cell’s transcriptional identity. Genome-wide mapping has identified thousands of bivalent promoters in human embryonic stem cells, most governing key developmental transcription factors.
Beyond DNA methylation and histone modifications, higher-order chromatin organization shapes the epigenome. Chromatin exists in two broad states: open euchromatin, which is accessible to the transcriptional machinery, and condensed heterochromatin, which is largely transcriptionally silent. Chromosomes are organized into topologically associating domains (TADs)—megabase-scale regions within which DNA sequences interact frequently with one another but rarely with sequences in neighboring TADs. The boundaries of TADs are anchored by the insulator protein CTCF and the cohesin complex, and disruption of these boundaries can lead to aberrant gene activation and disease.
The Epigenome in Development and Disease
Epigenomic reprogramming is one of the most dramatic events in mammalian biology. Shortly after fertilization, the embryo undergoes a global wave of DNA demethylation that erases most of the methylation patterns inherited from the parental gametes. A new methylation landscape is then laid down during implantation, and subsequent waves of epigenomic remodeling guide cell lineage specification throughout development. Genomic imprinting—the epigenomic silencing of one parental copy of a gene—survives this reprogramming and produces parent-of-origin-specific gene expression at roughly 100 loci in mammals.
Disruption of epigenomic regulation is a hallmark of cancer. Tumor cells typically show genome-wide hypomethylation, which can activate oncogenes and destabilize chromosomal structure, combined with focal hypermethylation at CpG islands of tumor suppressor genes. Mutations in genes encoding epigenomic regulators—including the DNA methyltransferase DNMT3A, the histone methyltransferase EZH2, and the chromatin remodeler ARID1A—are among the most frequent alterations observed across human cancers. These mutations reshape the epigenomic landscape of tumor cells and drive malignant transformation.
Epigenomic dysregulation also contributes to neurological disorders, metabolic diseases, and immune system dysfunction. In Alzheimer disease, specific DNA methylation changes at risk loci precede clinical symptoms by years. In type 2 diabetes, epigenomic alterations in pancreatic islet cells impair insulin secretion. Environmental exposures—including diet, tobacco smoke, air pollution, and psychosocial stress—can alter the epigenome in ways that persist long after the exposure ends, providing a molecular mechanism through which environment shapes phenotype and disease risk.
Mapping and Reading the Epigenome
Technologies for profiling the epigenome have advanced rapidly alongside DNA sequencing. Whole-genome bisulfite sequencing (WGBS) provides single-nucleotide-resolution maps of DNA methylation across the entire genome by treating DNA with sodium bisulfite, which converts unmethylated cytosines to uracil while leaving methylated cytosines intact. Reduced representation bisulfite sequencing (RRBS) offers a cost-effective alternative by enriching for CpG-dense regions. Array-based platforms, particularly the Illumina EPIC array, interrogate methylation at over 850,000 CpG sites and remain widely used in large-scale epidemiological studies.
Histone modifications are mapped using chromatin immunoprecipitation followed by sequencing (ChIP-seq), in which antibodies targeting a specific modification pull down the associated DNA fragments for sequencing. CUT&Tag and CUT&Run, more recently developed methods, achieve comparable resolution with far fewer cells and lower background noise. Chromatin accessibility is profiled by ATAC-seq, which uses a transposase to tag open chromatin regions, providing a genome-wide snapshot of the regulatory landscape. Single-cell versions of these assays now allow epigenomic profiling of individual cells, revealing heterogeneity that bulk methods obscure.
Bioinformatics tools integrate these diverse data types to annotate chromatin states genome-wide. ChromHMM and related algorithms use hidden Markov models to segment the genome into functional states—active promoters, enhancers, transcribed regions, repressed domains, and heterochromatin—based on the combinatorial patterns of histone marks. These chromatin state maps were central to the Roadmap Epigenomics and ENCODE projects and now serve as standard references for interpreting genomic variation in disease contexts.
The Epigenome in Medicine and Biotechnology
Epigenomic information is entering clinical practice through multiple routes. DNA methylation-based biomarkers are already used in oncology: the methylation status of the MGMT promoter predicts response to temozolomide chemotherapy in glioblastoma, and methylation-based liquid biopsy tests can detect multiple cancer types from a single blood draw by identifying tumor-specific methylation signatures in cell-free DNA. Epigenomic profiling is also being explored for classifying tumors, predicting treatment response, and monitoring minimal residual disease.
Epigenetic clocks—machine learning models trained on DNA methylation data from hundreds of CpG sites—estimate chronological and biological age with high accuracy. Steve Horvath developed the first pan-tissue clock in 2013 using methylation levels at 353 CpG sites. Second- and third-generation clocks have since been designed to predict not only chronological age but also mortality risk, the pace of biological ageing, and the effectiveness of anti-ageing interventions, making them valuable tools in personalized medicine and longevity research.
Pharmacologically, epigenomic regulators are established drug targets. DNA methyltransferase inhibitors (azacitidine and decitabine) and HDAC inhibitors (vorinostat and romidepsin) are approved for the treatment of hematologic malignancies. These drugs work by broadly resetting aberrant epigenomic marks in cancer cells, reactivating silenced tumor suppressor genes. Newer epigenomic therapies target more specific marks: EZH2 inhibitors for lymphoma, BET bromodomain inhibitors for cancers driven by aberrant enhancer activation, and IDH1/2 inhibitors that correct the production of an oncometabolite that disrupts histone and DNA demethylation.
Epigenome editing represents a more targeted approach. By fusing catalytically inactive CRISPR-Cas9 (dCas9) or zinc-finger proteins to epigenetic effector domains such as DNMT3A or KRAB, researchers can install or remove specific epigenomic marks at defined genomic locations without cutting the DNA. Unlike conventional genome editing, this strategy modifies gene expression while leaving the underlying sequence intact, reducing the risk of unintended mutations and chromosomal rearrangements associated with double-strand DNA breaks.
A 2024 study demonstrated that a single administration of lipid nanoparticles carrying epigenome editor mRNAs silenced the Pcsk9 gene in mouse liver cells for nearly a year, with the newly installed methylation marks persisting through liver regeneration. The first clinical trials of epigenome editing therapies are now under way, targeting conditions where durable gene silencing without permanent DNA changes is desirable.
Future Directions
Single-cell epigenomics is revealing the extent of epigenomic heterogeneity within tissues and tumors. Multi-omic approaches that simultaneously measure DNA methylation, histone modifications, chromatin accessibility, and transcriptomics in the same individual cell are beginning to connect epigenomic states directly to gene expression and cellular behavior. Spatial epigenomics methods, which preserve tissue architecture while profiling epigenomic marks, are adding another dimension by mapping where epigenomic variation occurs within intact organs.
Computational advances are accelerating epigenome interpretation. Deep learning models trained on large-scale epigenomic datasets can predict the functional impact of non-coding genetic variants on chromatin state and gene regulation—a capability that is critical for translating the results of genome-wide association studies into mechanistic understanding. Integration of epigenomic data with genotype information through approaches such as epigenome-wide Mendelian randomization is enabling causal inference about the role of specific epigenomic marks in disease.
As epigenome editing tools mature, the field is moving toward programmable control of gene expression for therapeutic purposes. Challenges remain in delivery, specificity, and ensuring the durability of epigenomic modifications in human tissues. Addressing the ethical dimensions of heritable epigenomic modifications—particularly in the context of germline interventions or transgenerational effects—will be as important as solving the technical ones. The epigenome occupies a unique position at the intersection of genetics, environment, and disease, and its continued study will reshape both basic biology and clinical medicine.
Frequently Asked Questions
Is the epigenome the same in every cell of the body? No. Although nearly every cell in the human body shares the same DNA sequence, different cell types carry distinct epigenomic patterns. These differences determine which genes are active in a given tissue—for example, the epigenome of a neuron silences muscle-specific genes while activating genes required for synaptic signaling. This cell-type-specific epigenomic landscape is established during development and maintained through cell division.
Can environmental factors change the epigenome? Yes. Diet, stress, chemical exposures, physical activity, and other environmental factors can alter DNA methylation patterns and histone modifications. Some of these changes are transient, while others persist for years or even across generations. Prenatal exposures, for instance, can reshape the epigenome of the developing fetus in ways that influence disease risk decades later.
Can epigenetic changes be passed from parents to children? In some cases, yes. While most epigenomic marks are erased and re-established between generations during embryonic reprogramming, certain marks—particularly at imprinted genes and some transposable elements—escape this erasure and are transmitted from parent to offspring. Environmental exposures such as famine or toxins have been associated with epigenomic changes detectable in subsequent generations in both animal models and human cohort studies, though the mechanisms and extent of transgenerational epigenetic inheritance in humans remain areas of active research.
Can the epigenome be edited for therapeutic purposes? Yes. Epigenome editing uses programmable DNA-binding proteins fused to epigenetic effector domains to add or remove specific chemical marks at targeted genomic locations. Unlike genome editing, epigenome editing does not cut or alter the DNA sequence itself. Preclinical studies have demonstrated durable gene silencing in animal models, and the first clinical trials of epigenome editing therapies are under way.
What are epigenetic clocks? Epigenetic clocks are machine learning models that estimate biological or chronological age from DNA methylation patterns at specific CpG sites across the genome. The original pan-tissue clock, developed by Steve Horvath in 2013, used methylation levels at 353 CpG sites to predict chronological age with high accuracy. Newer clocks have been trained to predict biological age, mortality risk, and the pace of ageing, making them valuable tools for evaluating anti-ageing interventions and disease prevention strategies.
Further Reading
Cell, The Mammalian Epigenome
Nature Reviews Genetics, Epigenetic Ageing Clocks: Statistical Methods and Emerging Computational Challenges
