| Mar 10, 2017 |
A three-dimensional map of the genome
|
|
(Nanowerk News) Cells face a daunting task. They have to neatly pack a several meter-long thread of genetic material into a nucleus that measures only five micrometers across. This origami creates spatial interactions between genes and their switches, which can affect human health and disease. Now, an international team of scientists has devised a powerful new technique that ‘maps’ this three-dimensional geography of the entire genome. Their paper is published in Nature ("Complex multi-enhancer contacts captured by Genome Architecture Mapping (GAM)").
|
 |
| These are colonies of mouse embryonic stem cells, where cell nuclei are stained in blue. The DNA from the nuclei is sequenced to infer the relative positions of genes and their switches.
|
|
Genes are activated to produce RNA and proteins, then switched off again when the molecules are no longer needed. Both the gene and its switches are DNA sequences, and they may lie far apart on the linear genome. This presents a challenge for the cell, because these regions usually have to be brought into contact to activate the gene.
|
|
It also creates a problem for scientists trying to understand one of the central questions in biology: how do cells decide which genes should be activated, and when? The answer will partly depend on matching every gene to its control sequences. But DNA strands are too thin to be tracked under the microscope, and even if that were possible, you’d have the vast amount of DNA in the nucleus to contend with. Imagine examining a tangle of yarn the size of the Earth in hopes of observing an encounter between individual strands.
|
Slicing nuclei and identifying contacts with the power of statistics
|
|
A new technique called Genome Architecture Mapping, or GAM, now helps to identify these contacts. It involves flash-freezing tissue or cells, then cutting thin slices of individual nuclei. The tiny amount of DNA within each slice of the nucleus is then sequenced, and the team deploys a mathematical model, named SLICE, to identify ‘hotspots’ of increased interaction between strands. The model looks at the frequency with which different genomic regions appear in the slice to infer information about the relative positions of genes and regions called enhancers that activate them.
|
|
“An analogy might be this; if you want to understand how school children interact you might take occasional photographs of where they sit in the canteen or appear together in the playground”, explains joint-lead author Ana Pombo, who began the project whilst working at the MRC London Institute of Medical Sciences (LMS) and is now based at the Berlin Institute for Medical Systems Biology, Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC) and the Berlin Institute of Health (BIH). “If you do that many times over a month, you will begin to see a pattern in those who often sit next to each other, or who run around together while playing. These random snapshots might tell you about their social interactions.”
|
|
“This is made possible by filtering out random encounters from real interactions using mathematical methods,” says the joint-lead author Mario Nicodemi at the Università di Napoli Federico II, who conceived such mathematical models and, aided by his PhD student Antonio Scialdone, developed them.
|
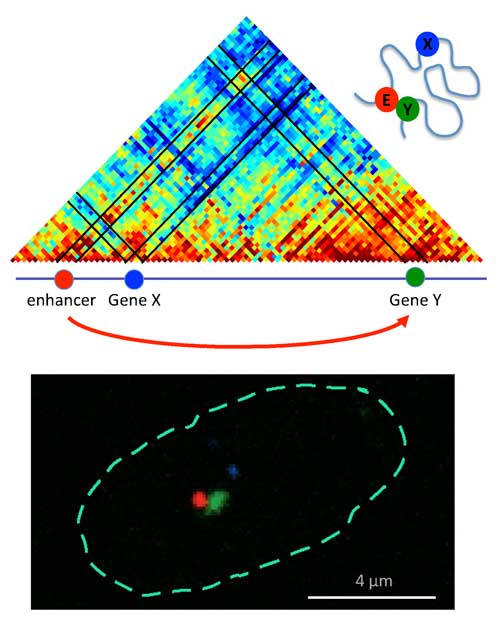 |
| Relative distance between genes. Top: Diagram with red coding for sequences that are close by, blue coding sequences further apart. Gene switch (enhancer, E) is close to gene Y but not gene X. Bottom: Position in the nucleus.
|
|
Paul Edwards, of the Hutchison/MRC Research Centre and Department of Pathology at the University of Cambridge, and Ana Pombo had the initial idea before the techniques necessary to do the experiment were available. “My research team optimised the approach, and as new technical steps came along we added them to our method,” she says.
|
Finding suspects linked to diseases
|
|
The study, which appears today in Nature, applies the method to mouse embryonic stem cells and the authors hope it will help shed light on many genes whose activity is disturbed in some very serious diseases. In some diseases, the problem lies within the sequence of a gene, but defects in regulatory regions found elsewhere in the genome can be equally dangerous and much harder to understand. The new data provides a long list of new suspects that can now be scrutinized by researchers.
|
|
Whilst previous studies have identified two-way contacts, this information does not reveal how often such contacts take place and by implication how important they might be, Pombo says: “They can spot that you and I are friends, but not how strong this friendship is relative to everyone else.”
|
|
“People have been measuring two-way contacts for a long time,” says Robert Beagrie, joint first author on the paper, who was a PhD student with Ana Pombo at the LMS when he collected the data for the study and is now based at the University of Oxford. “Those studies have often shown that you can have a set of different DNA elements that interact with each other in pairs. With this new approach we are able to generate a genome-wide catalogue of all the regions that we are confident interact in groups.” Now, the researchers are able to reliably detect and quantify so-called ‘three-way contacts’ in regions of the genome that are vigorously expressed.
|
|
Single cell technology: Great potential for the investigation of rare But perhaps the most notable advance of through GAM is that experiments are based on single cells – whether common or scarce in a tissue – and track their positions relative to each other within the tissue. Existing methods require lots of cells of the same type, which has made it difficult to study the biology and diseases of rare types. “There is huge potential for applying this in human tissue samples to catalogue contacts between regulatory regions and their target genes, and to use that to understand genetic variation and how it might alter aspects of nuclear biology,” Pombo says.
|
|
Some researchers are starting to show interest in using the technique to explore what happens when retroviruses insert their DNA into the genome of a host. Cancer scientists are also keen to create DNA maps of particular areas of a tumor. “By exploiting the unique nature of GAM data, mathematical models can reliably derive such information, opening the way to identify multiple, group interactions that could play a key role in the regulation of genes,” explains Nicodemi. “We can now ask whether a gene is contacted at the same time by all of its enhancers, or by each enhancer one at a time?”, Beagrie says. “We know that many genes that are important for early development have multiple enhancers. But how and why they are acting to regulate genes remain unanswered questions.”
|

