| Feb 13, 2020 |
Machine learning models accelerate the search for critical electrocatalysts |
| (Nanowerk Spotlight) Oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER) are among the core electrochemical processes in clean energy conversion and storage devices, such as metal-air batteries, water electrolyzers, and fuel cells. However, their reaction rates are inherently sluggish and the high cost and scarcity of state-of-the-art electrocatalysts such as palladium and platinum limit their large-scale and sustainable applications. |
| This has motivated researchers to develop stable, low-cost, and high-performance catalysts for these reactions. Single-atom catalysts (SACs), in which well-dispersed isolated metal atoms are anchored on appropriate substrates, have emerged as a new frontier of heterogeneous catalysts due to their highly increased coverage of active sites, enhanced catalytic performance, and maximal (100%) metal utilization. |
| In particular, metal-nitrogen-carbon (M-N-C) SACs, where a transition metal atom (M = Co, Fe, Ni, Mn, etc.) is located at the center of nitrogen (N) doped graphene support (C), show great promise as substitutes for precious metal electrocatalysts. |
| At least hundreds of M-N-C SACs exist due to a string of physical structural variables, such as different transition metals and various N/C combinations. Therefore, trial-and-error approaches are rather inadequate to search for highly efficient catalysts in a reasonable time scale. |
| Machine learning (ML) is an efficient statistical method, which builds models based on input data and computer algorithms to output the desired information. In recent years, ML has been applied to fast screening of materials with specific properties and catalysts with high performance. |
| Powerful ML algorithms may help find correlations between physical properties and limiting potentials of electrocatalytic materials. If successful, the discovery of electrocatalysts could be significantly accelerated. |
| So far, ML in catalysis is mainly focused on two aspects: 1) establishing the correlations of physical properties and adsorption strength of reaction intermediates; and 2) identifying the relationships between the intermediate adsorption strengths and the performance of the catalyst. However, the direct correlation between physical properties and limiting potentials has not been clearly studied. |
| In new work reported in Journal of Material Chemistry A ("Directly Predicting Limiting Potentials from Easily Obtainable Physical Properties of Graphene-Supported Single-Atom Electrocatalysts by Machine Learning"), Shiru Lin, Prof. Zhongfang Chen (Department of Chemistry, University of Puerto Rico, USA) and collaborators provide a new paradigm for directly predicting the catalytic performance from physical properties of catalyst candidates. |
| This work is the first to explore the correlation between physical properties and catalysis performance (limiting potentials) and applying this correlation to perform fast screening for similar graphene SACs. |
 |
| Figure 1. The schematic of three different aspects for catalyst development using machine learning techniques. (Reprinted with permission by Royal Society of Chemistry) |
| "By taking advantage of the ML algorithm and DFT computations, we depicted the underlying pattern of the physical properties of 104 graphene-supported SACs and their limiting potentials towards ORR/OER/HER reactions," Lin, first author of the paper, tells Nanowerk. "We further used the ML models for these three reactions to predict the catalytic performance of 260 other graphene-supported metal-nitrogen/carbon systems (M@NxCy)." |
| The researchers confirmed the reliability of their ML models by DFT-computed limiting potential values of the top ML-recommended electrocatalysts (0.61, 1.51, 0.003 V for ORR, OER, and HER, respectively). |
| The team selected three types of graphene-supported SACs as representatives of the M-N-C electrocatalysts (Figure 2), in which the central metal atom is 1) at the single vacancy with three carbon atoms (M@C3); 2) at the double vacancy with four nitrogen/carbon atoms (M@pyridine-N4 /M@C4); and 3) coordinated with four pyrrole nitrogen atoms (M@pyrrole-N4). 28 transition metals (except for Hg, La, and Ac) were employed as the central metal atoms. |
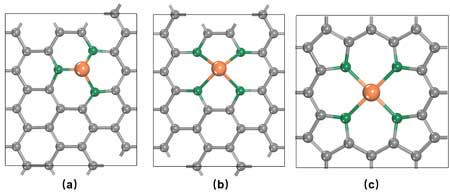 |
| Figure 2. Three types of graphene-supported M-N-C SACs. (Reprinted with permission by Royal Society of Chemistry) |
| "Focusing on intrinsic properties, we selected five features for ORR: the electron numbers of d orbital (d), the oxide formation enthalpy (Hof), Pauling electronegativity of the central metal atom (Em), the sum of Pauling electronegativity of surrounding atoms (Es), and the average of the pKa values of the surrounding atoms," Lin explains. |
| "Hof firmly connects with the ability of the metal atom to react with oxygen, which can be obtained by the energy difference between the experimentally determined cohesive energy of the bulk metal, and the formation enthalpy of the metal's most stable oxide," he adds. |
| He points out that the average of the pKa values of the surrounding atoms is introduced for the first time as a feature for catalytic activity to distinguish pyridine-N and pyrrole-N; C6-C and C5-C surrounding atoms. |
| In the OER model, similar to oxide formation enthalpy, hydride formation enthalpy was introduced using the most stable metal hydrides (including both 2D and 3D metal hydrides). In HER, Hof was excluded, while other features were all included. |
| All three ML models achieved excellent performances: the training and test scores are all higher than 0.90, and their mean square errors are rather low (see Figure 3). |
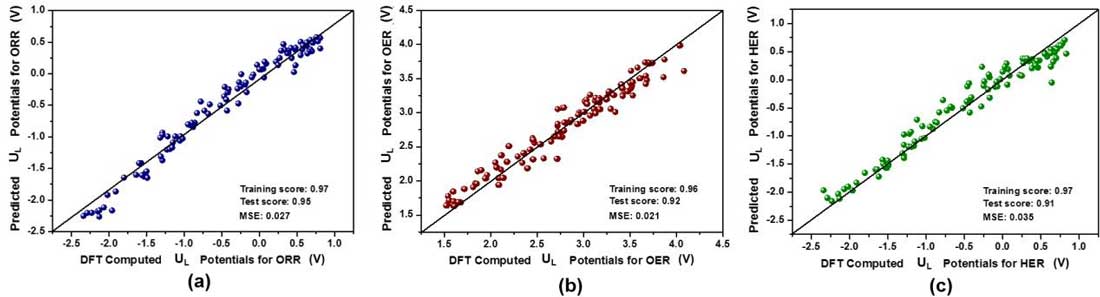 |
| Figure 3. Comparison between DFT and ML predicted limiting potential (UL) values of (a) ORR, (b) OER, and (c) HER, where both training and testing data points are presented. (Reprinted with permission by Royal Society of Chemistry) (click on image to enlarge) |
| "To encode the significance of different properties towards UL, we compared the importance of these features," Lin notes. "For the three reactions, the number of electrons has the largest feature importance, followed by the oxygen formation enthalpy for ORR, hydride formation enthalpy for OER, while the electronegativity of central atoms is in second place for HER." |
| The team expects that the results of their work will shed new light on using machine learning applications for screening electrocatalysts, as well as the development of single atom catalysts for energy-related electrocatalytic reactions. This work shows that combining ML and DFT computations is very effective and can significantly accelerate the screening process. |
| "By utilizing only a few easily available intrinsic physical features of metal-nitrogen-carbons, our machine learning models can well predict the limiting potentials of graphene-supported SACs towards ORR/OER/HER," adds Professor Chen. "Without any geometry optimization, total energy calculation, or examining reaction pathways, our designed ML process can dramatically narrow down the candidate list of metal-nitrogen-carbon single-atom catalysts." |
| "Our strategy can be used to screen and design other electrochemical catalysts towards for example nitrogen reduction and CO2 reduction reactions," he concludes. "Directly predicting catalytic performance of electrocatalysts from the easily obtainable parameters of catalysts is giving us a revolutionary approach for future catalysts design and will dramatically accelerate the discovery of more efficient catalysts towards important chemical processes in the very near future." |
 By
Michael
Berger
– Michael is author of four books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Technology (2009),
Nanotechnology: The Future is Tiny (2016),
Nanoengineering: The Skills and Tools Making Technology Invisible (2019), and
Waste not! How Nanotechnologies Can Increase Efficiencies Throughout Society (2025)
Copyright ©
Nanowerk LLC
By
Michael
Berger
– Michael is author of four books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Technology (2009),
Nanotechnology: The Future is Tiny (2016),
Nanoengineering: The Skills and Tools Making Technology Invisible (2019), and
Waste not! How Nanotechnologies Can Increase Efficiencies Throughout Society (2025)
Copyright ©
Nanowerk LLC
|

Become a Spotlight guest author! Join our large and growing group of guest contributors. Have you just published a scientific paper or have other exciting developments to share with the nanotechnology community? Here is how to publish on nanowerk.com. |
