| Dec 10, 2023 |
Accelerating the discovery of organic electronic materials through data-driven molecular design |
| (Nanowerk Spotlight) The pursuit of more efficient organic electronics, from OLED displays to organic solar cells, has been an enduring challenge across materials science. But the vastness of possible organic compounds and limitations of virtual screening methods – which are advanced computational techniques used to evaluate and predict the properties of materials – have hindered the pace of novel materials discovery. |
| Researchers have strived to systematically navigate chemical space and accurately predict molecular properties to unlock new high-performing compounds. However, existing approaches bias the exploration toward incremental improvements or struggle to extract generative design principles. |
| On one hand, bottom-up strategies start with known molecular building blocks and combine them in different ways, enabling rapid virtual screening. However, they are biased toward incremental improvements of existing materials rather than wholly new families of compounds. On the other hand, top-down data mining of structural databases casts a wider net but struggles to extract actionable design guidelines from the outputs. |
| In the field of materials science, there are two main strategies for discovering new materials. On one hand, 'bottom-up' strategies begin with known molecular building blocks. Think of it like using LEGO bricks to create new structures; scientists combine these blocks in various ways, enabling rapid computerized testing of their properties. This method, however, tends to focus on making small improvements to existing materials rather than creating completely new ones. On the other hand, 'top-down' data mining involves searching through vast databases of molecular structures to find potential new materials. It's akin to casting a wide net to see what you can find, but this approach often struggles to identify practical guidelines for creating these materials from the data it uncovers. |
| Meanwhile, high computational costs constrain both approaches from exploring molecular spaces as vast as nature itself. Predictive machine learning models have emerged as a way to bypass expensive quantum chemistry calculations. But model development is hampered by the scarcity of sizable, high-quality training datasets with relevant excited state properties calculated from first principles. |
| Now, researchers from EPFL have married bottom-up construction with top-down data mining in a hybrid materials discovery platform. The enabling breakthroughs are a curated dataset of 117,000 synthesized organic structures with TDDFT-computed properties, automated identification of cross-coupling sites to generate new compounds on the fly, and fast yet accurate machine learning models to predict key optical characteristics. |
| The findings have been published in Advanced Materials ("Data-Driven Discovery of Organic Electronic Materials Enabled by Hybrid Top-Down/Bottom-Up Design"). |
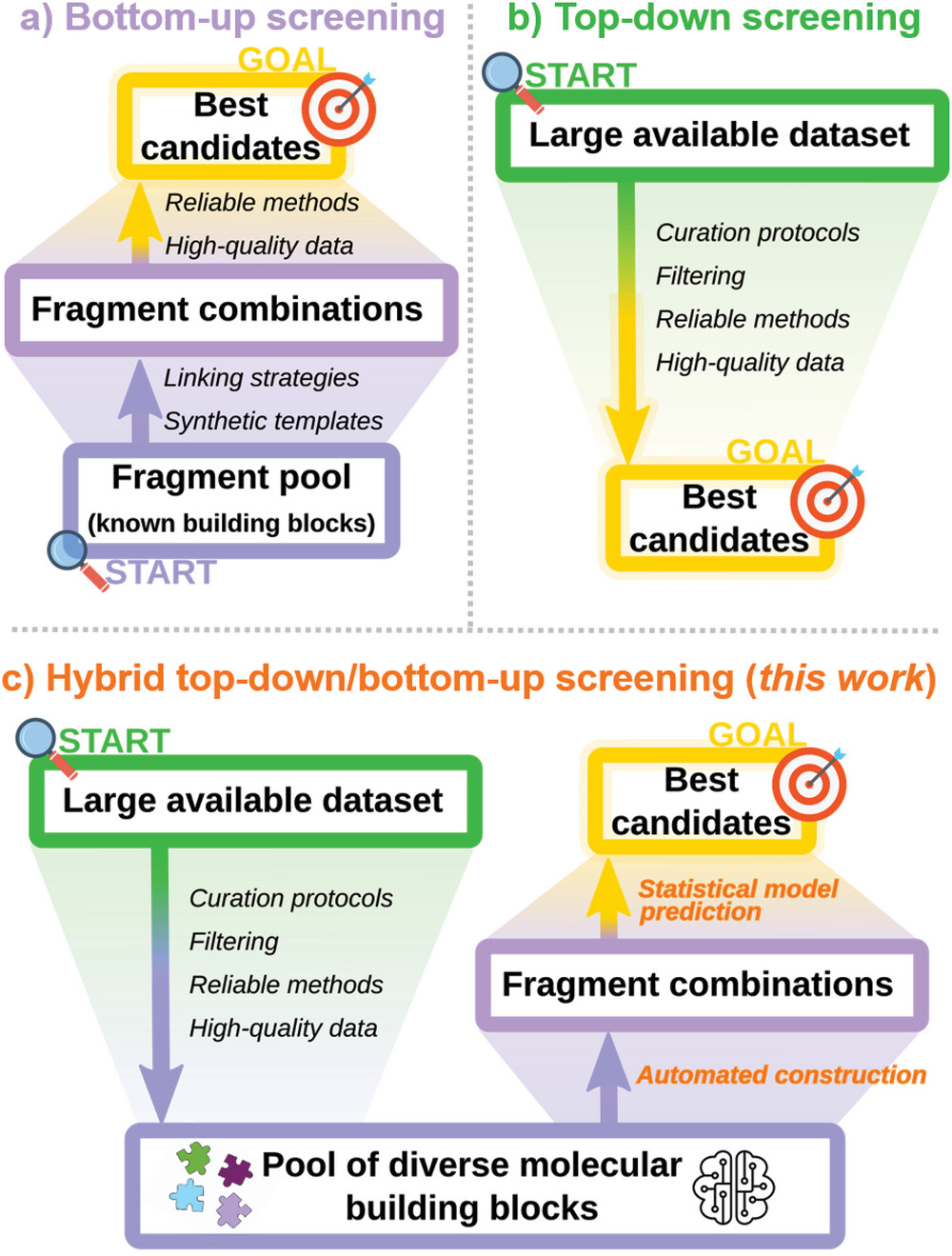 |
| General view of the a) bottom-up (purple) and b) top-down (green) philosophies of high-throughput virtual screening, with with end-goals in yellow. Requirements for the different stages of a successful screening campaign are indicated in italics and datasets are shown as blocks, whose widths represent the datasets’ sizes relative to one another. c) The hybrid screening methodology described in this work, with the union of top-down (green) and bottom-up (purple) elements enabled by statistical models and automated fragment coupling (orange). (© Wiley-VCH) |
| Powered by this data-driven infrastructure, the researchers demonstrated ultra-large-scale screening for singlet fission candidates. From over a million constructed donor-acceptor dimers, hundreds of thousands were flagged as having ideal excited state energetics for solar cells. Many feature unusual molecular motifs like nitrones and furoxans that have scarcely been investigated for singlet fission till now. |
| Tapping the full diversity of already synthesized and stable compounds debiases the search from conventional building blocks. And by training models on quantum-accurate data spanning known chemical space, reliable predictions unlock virtual screening at unprecedented scales. |
| The researchers extracted over 167,000 crystal structures from the Cambridge Structural Database, representing the largest public compilation of characterized synthetic compounds. Extensive curation eliminated duplicates, errors, and nonsensically bonded moieties. |
| The final filtered set of 117K compounds provides a structurally diverse palette spanning known organic chemistry far beyond conventional molecular datasets. Compared even to the 134K small molecules in the established QM9 set, this covers more elements across a greater range of sizes without constraints. |
| From each refined crystal structure, a suite of electronic properties was calculated using density functional approximations. This includes frontier orbital energies, multiple low-lying singlet and triplet excitation levels, and quantitative descriptors of transition character. Together these properties inform exciton behavior and charge transfer character, underlying applications from OLED emitters to organic photovoltaics. |
| To transform static molecule snapshots into versatile building blocks, the researchers developed a topology-based tool that flags potential carbon sites suitable for cross-coupling reactions. Focusing on unsaturated C-H positions, they enumerate the local chemical environments to identify symmetry-equivalent groups. |
| This protocol mimics retrosynthetic analysis to decode sites where fragments could be stitched together by established carbon-carbon bond formations. Codifying the ability to couple directly extracts synthesis-relevant information from 3D structural data. It also enables algorithmic assembly of derivatives not initially present in the dataset. |
| To bypass the need for expensive excited state calculations on every new hypothetical compound, the team trained machine learning models using the precomputed data. The inputs representation incorporated geometry-dependent London and Axilrod-Teller-Muto potentials—physics-inspired atomic pairwise and triplet terms. |
| The models achieved remarkable accuracy in predicting singlet and triplet excitation energies for diverse organic structures. This required adding several thousand computer-generated dimers to the training data for sufficient representation of bonded molecular pairs. The models recapitulate quantum chemistry with a fraction of the computational expense, forecasting optical properties in a flash. |
| The researchers demonstrated the capabilities of their hybrid discovery infrastructure by constructing and screening over one million donor-acceptor dimers for intramolecular singlet fission candidacy. Known singlet fission materials are scarce, sensitive, suffer from low triplet energy, and derive mostly from oligoacene derivatives. |
| By stipulating complementary design criteria based on frontier orbital patterns and excitation energetics, the team rationally selected donor and acceptor fragments with appropriate characteristics from within their molecular database. Automated coupling of these thousands of building blocks followed by ML-accelerated screening yielded almost 560,000 structurally distinct dimers with ideal thermodynamic signatures for singlet fission and triplet energies suited for common photovoltaic absorbers like silicon and CdTe. |
| Many contain rarely investigated acceptor groups like nitrones and furoxans that hold untapped promise for singlet fission, as corroborated through substructure analysis. Starting directly from molecules already successfully synthesized avoids reliance on conventional intuitions about productive chemical subunits. This highlights how data-driven compound generation powered by predictive modeling provides an agnostic avenue to uncharted molecular territories with desired properties. |
 By
Michael
Berger
– Michael is author of four books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Technology (2009),
Nanotechnology: The Future is Tiny (2016),
Nanoengineering: The Skills and Tools Making Technology Invisible (2019), and
Waste not! How Nanotechnologies Can Increase Efficiencies Throughout Society (2025)
Copyright ©
Nanowerk LLC
By
Michael
Berger
– Michael is author of four books by the Royal Society of Chemistry:
Nano-Society: Pushing the Boundaries of Technology (2009),
Nanotechnology: The Future is Tiny (2016),
Nanoengineering: The Skills and Tools Making Technology Invisible (2019), and
Waste not! How Nanotechnologies Can Increase Efficiencies Throughout Society (2025)
Copyright ©
Nanowerk LLC
|

Become a Spotlight guest author! Join our large and growing group of guest contributors. Have you just published a scientific paper or have other exciting developments to share with the nanotechnology community? Here is how to publish on nanowerk.com. |
