Metagenomics: How It Works, Core Technologies, and Applications
What Is Metagenomics?
Metagenomics is the culture-independent genomic analysis of the collective DNA recovered directly from an environmental or clinical sample. Rather than isolating and growing individual organisms in the laboratory, metagenomics extracts and sequences all of the genetic material present in a sample at once—soil, seawater, human stool, a hospital air filter—capturing the genomes of bacteria, archaea, viruses, fungi, and microbial eukaryotes in a single experiment. The approach provides both a taxonomic census (which organisms are present) and a functional inventory (which genes and metabolic pathways they encode), making it one of the most powerful tools in modern microbial ecology and molecular biology.
The term was coined by Jo Handelsman and colleagues in 1998 to describe the application of genomic methods to the total DNA—the metagenome—of a microbial community. The concept emerged from a long-standing recognition that standard laboratory culture captures only a small fraction of microbial diversity: in many environments, as many as 99% of microorganisms resist cultivation under standard conditions.
Early metagenomic studies in the 2000s relied on cloning environmental DNA into bacterial hosts and screening the resulting libraries for functions of interest. The landmark 2004 shotgun sequencing of microbial populations in the Sargasso Sea by J. Craig Venter and colleagues, which generated over one billion base pairs and identified more than 1,800 genomic species, demonstrated that high-throughput DNA sequencing could be applied directly to complex natural communities at enormous scale.
Since then, the combination of steadily declining sequencing costs, increasingly powerful computational methods, and the proliferation of reference databases has transformed metagenomics from a niche research technique into a routine tool across biomedicine, agriculture, environmental monitoring, and industrial biotechnology. Large-scale cataloging efforts such as the Earth Microbiome Project and the Integrated Microbial Genomes (IMG/M) platform have assembled reference datasets containing tens of thousands of metagenome-assembled genomes, providing a scaffold against which new metagenomic datasets can be interpreted.
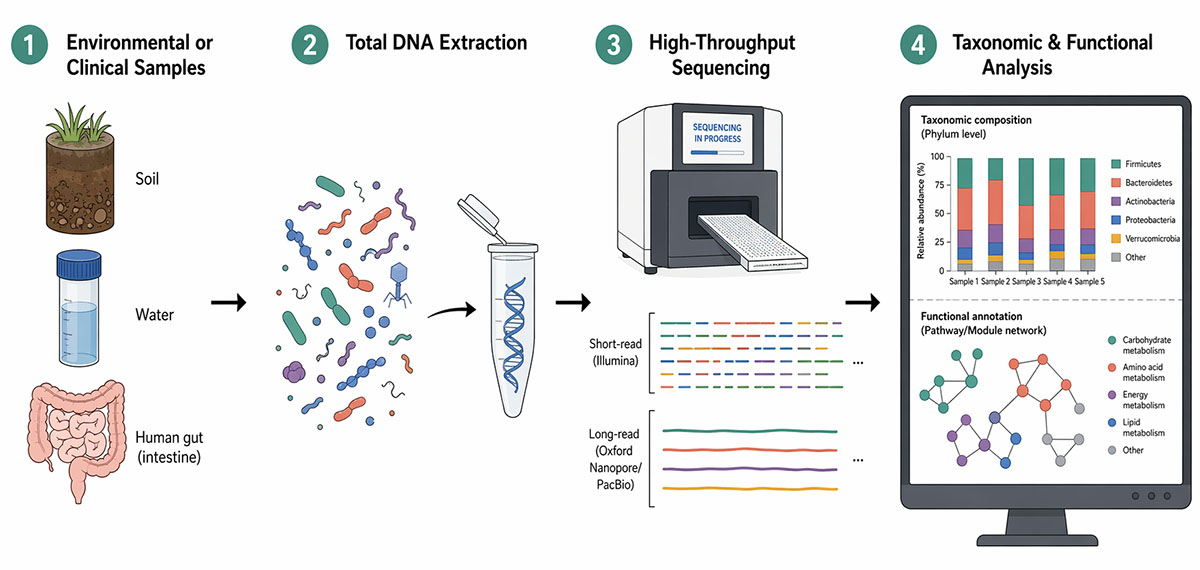
How Does Metagenomics Work?
A metagenomic experiment begins with sample collection and DNA extraction. The choice of extraction protocol is critical because different methods recover DNA from different organisms with varying efficiency; harsh lysis conditions may be needed to break open thick-walled bacterial spores or fungal cells, while gentle protocols preserve high-molecular-weight DNA suitable for long-read sequencing. Once purified, the DNA is processed into sequencing libraries. Two fundamentally different strategies exist: amplicon-based approaches and whole-genome shotgun (WGS) metagenomics.
Amplicon-based metagenomics targets a specific marker gene—most commonly the 16S ribosomal RNA (rRNA) gene for bacteria and archaea, or the internal transcribed spacer (ITS) region for fungi. The marker gene is amplified by polymerase chain reaction (PCR) using conserved primers that flank a variable region, and the resulting amplicons are sequenced. Comparison of the variable sequences against curated reference databases (such as SILVA or the Ribosomal Database Project) allows taxonomic classification of each sequence read. Amplicon surveys are cost-effective and well-established, but they are limited to detecting organisms with the targeted marker gene and provide no direct information about the functional capabilities of the community.
Whole-genome shotgun metagenomics sequences all DNA in the extract without prior amplification of a specific gene. The extracted DNA is fragmented, adapter-ligated, and sequenced on high-throughput platforms. Short-read technologies such as Illumina produce hundreds of millions of paired-end reads of 150 to 300 nucleotides, providing deep coverage of the community at relatively low cost per base.
Long-read platforms from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies generate reads of 10,000 to more than 100,000 nucleotides, which span repetitive regions and enable more contiguous genome assemblies. Nanopore devices such as the MinION additionally offer real-time data streaming and portable form factors suited to field-based metagenomics in remote environments or clinical point-of-care settings. Many current studies combine both short-read and long-read technologies in a hybrid assembly strategy: short reads supply accuracy and depth, while long reads resolve structural complexity.
After sequencing, raw reads undergo quality control—adapter trimming, removal of low-quality bases, and, for host-associated samples, filtering of host DNA. The cleaned reads are then analyzed through two complementary strategies. Read-based approaches align individual reads or read pairs directly against reference databases to assign taxonomy and annotate gene function, using tools such as Kraken2, MetaPhlAn, and HUMAnN.
Assembly-based approaches first reconstruct longer contiguous sequences (contigs) from overlapping reads, then group contigs from the same organism into metagenome-assembled genomes (MAGs) through a process known as binning. MAGs can approach near-complete genomes of individual species, even for organisms that have never been grown in culture, enabling detailed metabolic reconstruction and comparative genomics.
Beyond DNA, metatranscriptomics (sequencing of community RNA) and metaproteomics (mass-spectrometric analysis of community proteins) extend the metagenomic framework from what organisms can do (gene content) to what they are doing at a given moment (gene expression and protein production). Integrating these multi-omic layers provides a more dynamic picture of microbial community function than DNA-based metagenomics alone.
Comparison of Metagenomic Approaches
The choice between amplicon sequencing, short-read shotgun metagenomics, and long-read shotgun metagenomics involves trade-offs in cost, resolution, and the types of biological questions each approach can address. The table below summarizes these differences.
| Feature | 16S/ITS Amplicon Sequencing | Short-Read Shotgun Metagenomics | Long-Read Shotgun Metagenomics |
|---|---|---|---|
| Target | Single marker gene (16S rRNA, ITS) | All DNA in sample | All DNA in sample |
| Taxonomic scope | Bacteria and archaea (16S) or fungi (ITS) | All domains of life plus viruses | All domains of life plus viruses |
| Taxonomic resolution | Genus to species (variable region dependent) | Species to strain | Species to strain (improved for closely related taxa) |
| Functional information | Inferred only (e.g., PICRUSt predictions) | Direct gene and pathway annotation | Direct gene and pathway annotation with better operon resolution |
| Genome assembly | Not applicable | Fragmented MAGs typical | Near-complete MAGs, including plasmids and mobile elements |
| Relative cost per sample | Low | Moderate to high | High |
For large-scale ecological surveys in which the primary goal is community composition across hundreds of samples, amplicon sequencing remains the most practical choice. When functional annotation, strain-level resolution, or the discovery of novel organisms is the objective, shotgun metagenomics is essential. Long-read sequencing adds the most value when complete or near-complete genome recovery is needed—for example, to resolve mobile genetic elements carrying antibiotic resistance genes or to link specific metabolic pathways unambiguously to their host organism.
Applications in Research and Medicine
Metagenomics has reshaped the study of the human microbiome. The Human Microbiome Project and subsequent large-cohort studies used metagenomic sequencing to catalogue the microbial communities inhabiting the gut, skin, oral cavity, and urogenital tract, revealing that the human body harbors trillions of microbial cells whose collective gene catalogue dwarfs the human genome. Metagenomic associations between gut microbiome composition and conditions such as inflammatory bowel disease, type 2 diabetes, obesity, colorectal cancer, and responses to cancer immunotherapy have opened new avenues for diagnostics and therapeutics, including fecal microbiota transplantation and rationally designed microbial consortia.
Clinical metagenomics—the direct sequencing of patient samples to identify infectious agents—is one of the most rapidly advancing applications. Metagenomic next-generation sequencing (mNGS) can detect bacteria, viruses, fungi, and parasites in a single assay without requiring clinicians to specify which pathogen to test for, making it particularly valuable for cases of unexplained encephalitis, sepsis, or pneumonia where conventional culture and targeted molecular tests return negative results. mNGS simultaneously provides information on antimicrobial resistance genes and, when RNA is sequenced, on the host immune response. Several clinical laboratories now offer validated mNGS assays, although challenges around turnaround time, cost, standardization, and distinguishing colonizers from true pathogens remain areas of active development.
In environmental science, metagenomics has unveiled the staggering diversity of microbial life in soils, oceans, freshwater, and extreme environments such as deep-sea hydrothermal vents and permafrost. The Tara Oceans expedition, for instance, applied shotgun metagenomics to samples collected across all major ocean basins, revealing millions of previously unknown genes and new phyla of bacteria and archaea. Agricultural applications include profiling soil microbiomes to understand nutrient cycling and plant health, monitoring rumen communities to improve livestock feed efficiency, and tracking antimicrobial resistance genes along the farm-to-fork continuum.
Industrial biotechnology exploits metagenomics as a source of novel enzymes. By screening metagenomic libraries or mining assembled sequence data, researchers have identified thermostable cellulases, lipases, and xylanases from extreme environments that can be harnessed for biofuel production, textile processing, and pharmaceutical synthesis. The ability to access the genetic repertoire of uncultured organisms gives metagenomics a significant advantage over traditional enzyme discovery, which is limited to the small fraction of organisms that grow in the laboratory.
Limitations and Challenges
Despite its power, metagenomics faces several persistent technical and interpretive challenges. DNA extraction bias is a fundamental concern: no single protocol recovers DNA from all organism types with equal efficiency, meaning that the observed community composition is always filtered through the extraction method. Gram-positive bacteria with thick peptidoglycan walls, bacterial endospores, and organisms embedded in biofilms or host tissue may be under-represented if extraction protocols do not include sufficient mechanical or chemical lysis.
Computational challenges scale with data volume. Assembling genomes from complex communities such as soil, which can contain thousands of species per gram, remains difficult because of the uneven abundance distribution of community members. Low-abundance organisms generate too few reads for reliable assembly, while highly similar genomes from closely related species produce chimeric contigs. Binning algorithms that assign contigs to MAGs have improved substantially with machine-learning approaches, but validation of MAG quality still relies on indirect metrics such as completeness and contamination estimates from marker gene sets.
A significant proportion of genes identified in metagenomic datasets have no match in existing reference databases—the so-called biological dark matter. For some environments, 40 to 70% of predicted open reading frames cannot be assigned a function. This gap limits the ability of metagenomics to move beyond cataloging genes toward understanding community-level metabolism. Expanding and curating reference databases, particularly for undersampled environments and non-model organisms, remains a priority for the field.
For clinical applications, distinguishing true pathogens from commensals, contaminants, and background environmental DNA is a non-trivial problem. Metagenomic sequencing detects DNA from all sources, including dead organisms and free DNA fragments, and the presence of a microbial genome in a clinical sample does not necessarily indicate active infection. Integrating metagenomic data with clinical metadata, host transcriptomic signatures, and quantitative abundance thresholds is an emerging strategy to improve diagnostic specificity.
Future Directions
Several converging trends are expanding the scope of metagenomics. Long-read sequencing at reduced cost is making complete or near-complete genome recovery routine rather than exceptional, enabling the linkage of mobile genetic elements—such as plasmids carrying antibiotic resistance or phage genomes—to their host organisms at a resolution that short reads alone cannot achieve. Spatial metagenomics, which combines metagenomic sequencing with spatially resolved sampling or in situ hybridization, is beginning to map microbial communities at micrometer scales within tissues, biofilms, and soil aggregates.
Artificial intelligence and machine learning are reshaping metagenomic data analysis. Deep learning models trained on large reference genome collections improve taxonomic classification of novel sequences, predict gene function from protein structure rather than sequence homology, and detect disease-associated microbial signatures in clinical datasets. Protein structure prediction tools such as AlphaFold are being applied to the millions of uncharacterized metagenomic protein sequences, offering a path toward functional annotation of the metagenomic dark matter that sequence-based methods cannot reach.
Integration of metagenomics with other omics layers—transcriptomics, proteomics, metabolomics, and host genomics—is moving the field from descriptive cataloging toward mechanistic understanding of how microbial communities interact with their environments and hosts. As sequencing technologies become cheaper, faster, and more portable, and as computational tools grow more sophisticated, metagenomics is poised to become a routine component of personalized medicine, environmental surveillance, and agricultural management.
Frequently Asked Questions
What is the difference between metagenomics and genomics? Genomics studies the complete genetic material of a single organism, typically after isolating and culturing it in the laboratory. Metagenomics bypasses cultivation entirely and sequences all of the DNA extracted from an environmental or clinical sample at once, capturing genetic material from hundreds or thousands of species simultaneously. This culture-independent approach reveals organisms that cannot be grown under standard laboratory conditions, which represent the vast majority of microbial life on Earth.
Can metagenomics identify viruses as well as bacteria? Yes. Shotgun metagenomic sequencing captures all DNA and, when RNA is also extracted and reverse-transcribed, all RNA present in a sample, including viral genomes. Because viruses lack a universal marker gene equivalent to the 16S rRNA gene used for bacteria, shotgun metagenomics is often the only culture-independent method capable of detecting novel or highly divergent viruses. Clinical metagenomic sequencing has been used to identify previously unknown viral pathogens in patients with unexplained infections.
How much does a metagenomic sequencing experiment cost? Costs vary widely depending on sample type, sequencing depth, and platform. A typical amplicon-based 16S rRNA survey of a few hundred samples can cost a few thousand dollars in sequencing reagents alone. Whole-genome shotgun metagenomics is more expensive per sample because it requires deeper sequencing to capture low-abundance organisms and assemble genomes. Long-read sequencing on platforms such as PacBio or Oxford Nanopore adds further cost but resolves repetitive genomic regions that short reads cannot. Computational infrastructure for data analysis represents an additional and often underestimated expense.
What is a metagenome-assembled genome (MAG)? A metagenome-assembled genome is a draft genome of a single organism reconstructed computationally from a complex metagenomic dataset. After shotgun sequencing reads are assembled into longer contiguous sequences (contigs), binning algorithms group contigs that likely originate from the same organism based on features such as nucleotide composition and read coverage depth. A high-quality MAG typically captures more than 90% of a genome with less than 5% contamination from other organisms. MAGs have been critical for characterizing microbes that have never been grown in the laboratory, expanding the known tree of life by thousands of species.
What is the difference between metagenomics and metatranscriptomics? Metagenomics sequences the total DNA in a sample and reveals which organisms are present and which genes they carry. Metatranscriptomics sequences the total RNA (after converting it to complementary DNA) and reveals which genes are actively being expressed at the time of sampling. DNA-based metagenomics provides a relatively stable snapshot of genetic potential, while RNA-based metatranscriptomics captures dynamic gene activity that changes with environmental conditions, growth phase, or host interactions. The two approaches are complementary and are often used together in multi-omic study designs.
Further Reading
Annual Review of Genetics, Metagenomics: Genomic Analysis of Microbial Communities
Nature Reviews Genetics, Clinical Metagenomics
Nature Reviews Genetics, Sequencing-Based Analysis of Microbiomes
Nature Reviews Methods Primers, Analysis of Metagenomic Data
