Protein Folding: Definition, Mechanism, Misfolding, and AlphaFold
What Is Protein Folding?
Definition: Protein folding is the physical process by which a linear chain of amino acids adopts, often with cellular assistance, the three-dimensional structure or ensemble of structures required for biological function.
Protein folding is the process by which a newly made polypeptide, a linear chain of amino acids joined end to end, collapses and rearranges into the compact, precisely organized three-dimensional structure that allows it to work. The shape a protein settles into is not incidental: an enzyme can only catalyze a reaction, an antibody can only recognize its target, and a transport protein can only carry its cargo when its chain is folded into the correct arrangement. In molecular biology, the link between structure and function is so tight that, for many proteins, folding is the step that turns an amino acid sequence into a working molecular machine rather than a flexible chain of building blocks.
The scale of the problem is what makes folding remarkable. A modest protein of 100 residues, with only a handful of possible orientations at each position, could in principle adopt an astronomically large number of conformations, on the order of 1047 or more. Sampling them all at random would take far longer than the age of the universe, an observation known as Levinthal's paradox. Yet small proteins routinely reach their correct shape in microseconds to seconds. The resolution is that folding is not a blind search but a strongly biased, downhill process guided by the physics of the chain and its environment.
At a glance:
- Protein folding: a polypeptide chain adopting its functional 3D structure or ensemble
- Driven by: the hydrophobic effect, hydrogen bonds, van der Waals forces, electrostatics and disulfide bonds
- Information source: largely encoded in the amino acid sequence (Anfinsen's dogma)
- In the cell: assisted by molecular chaperones to prevent aggregation
- When it fails: misfolding and aggregation are implicated in Alzheimer, Parkinson and other diseases
- Predicting it: AlphaFold models many well-ordered protein structures from sequence, with important caveats
Folding is also fragile. When a chain fails to reach its native shape, the result is not simply an inactive molecule but often a sticky, aggregation-prone species that can damage cells. Because of this, folding sits at the center of fields ranging from structural protein science and neurodegeneration research to biopharmaceutical manufacturing, where a misfolded therapeutic protein is a failed product.
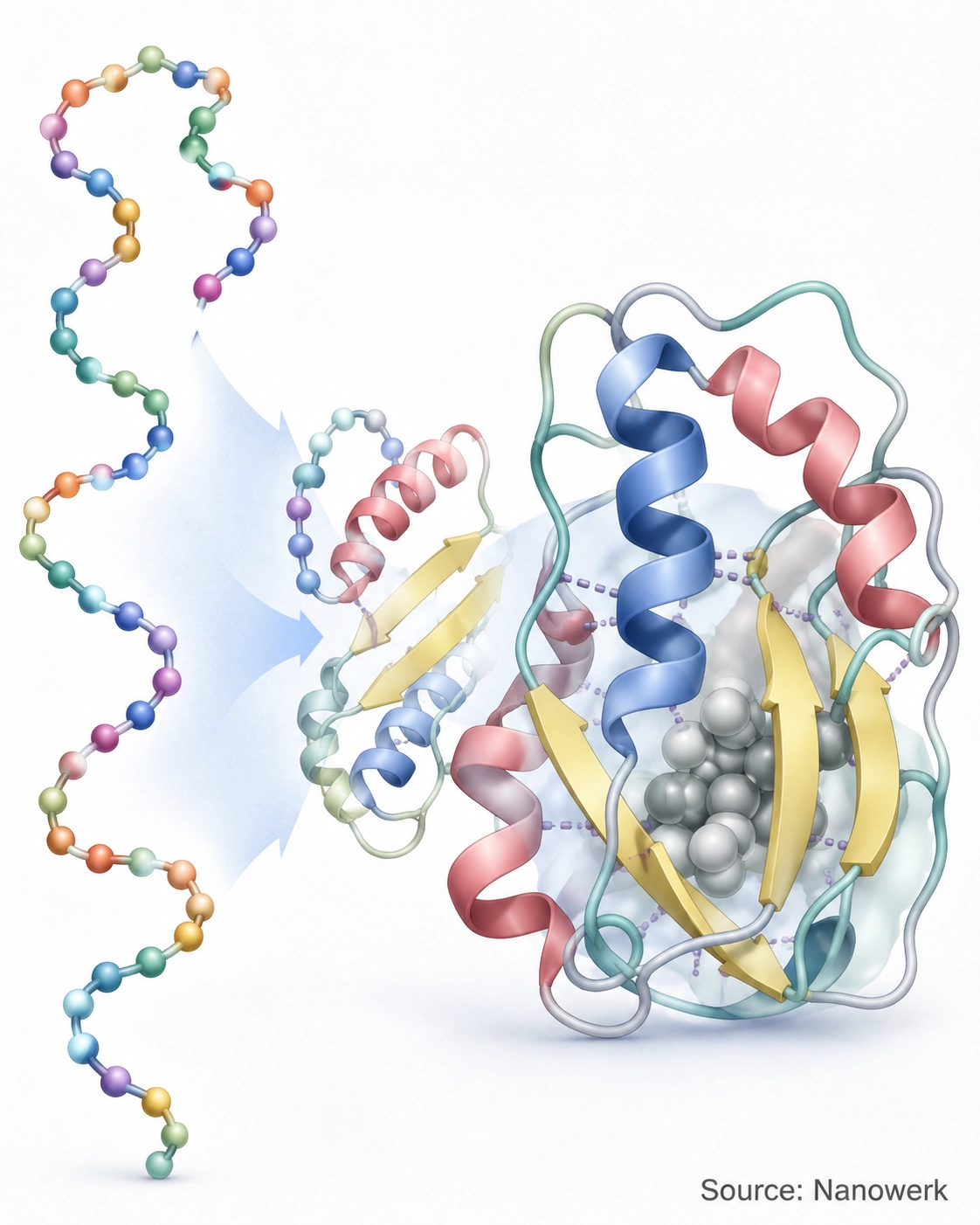
The Four Levels of Protein Structure
A folded protein is described at four hierarchical levels, and folding is essentially the journey from the first level to the higher ones. The primary structure is the linear order of amino acids, set during protein synthesis when the ribosome reads messenger RNA. The secondary structure consists of local, regularly repeating patterns, principally the alpha helix and the beta sheet, stabilized by hydrogen bonds along the backbone. The tertiary structure is the overall three-dimensional fold of a single chain, and the quaternary structure is the assembly of several folded chains into one functional complex, as in hemoglobin's four subunits.
These levels are not just a descriptive convenience; they map onto the order in which folding tends to happen. Local secondary structure can form within nanoseconds, while the long-range contacts that define the tertiary fold take longer to settle. The table below summarizes the four levels and the interactions that hold each one together.
| Structural level | What it describes | Main stabilizing interactions |
|---|---|---|
| Primary | The linear amino acid sequence of the chain | Covalent peptide bonds |
| Secondary | Local motifs: alpha helices and beta sheets | Backbone hydrogen bonds |
| Tertiary | The full 3D fold of one polypeptide chain | Hydrophobic effect, hydrogen bonds, salt bridges, disulfide bonds |
| Quaternary | Assembly of multiple folded chains into a complex | The same non-covalent forces acting between subunits |
A point worth drawing out from this comparison is that, apart from covalent crosslinks such as disulfide bonds, the higher levels of protein structure are maintained mostly by many individually weak, reversible interactions. That fragility is the price of function: a structure stable enough to work but flexible enough to change shape, bind partners and be taken apart and recycled when no longer needed.
How Proteins Fold: Driving Forces and the Energy Landscape
The single most important force in folding is the hydrophobic effect. Many amino acids carry nonpolar side chains, and in the watery interior of a cell these groups are energetically favored when they cluster away from the solvent, reducing the ordering of water molecules around them. As the chain collapses, hydrophobic residues are buried in a tightly packed core while polar and charged residues remain on the surface in contact with water. This burial provides much of the driving force for compaction and largely sets the protein's overall architecture.
Folding is then refined by several weaker interactions acting together. Hydrogen bonds along the backbone lock in helices and sheets; van der Waals contacts let buried side chains pack against one another with almost crystalline efficiency; electrostatic attractions, often called salt bridges, form between oppositely charged groups; and in many secreted proteins covalent disulfide bonds between cysteine residues clamp distant parts of the chain together. None of these is individually strong, but summed over an entire protein they specify one structure far more stable than the alternatives.
A useful way to picture the process is the folding funnel. Instead of a single fixed route, an unfolded chain can begin from many starting conformations, each high in free energy. As favorable contacts form, the chain moves down the sloping walls of a funnel-shaped energy landscape toward the native state at its bottom. Many paths lead to the same destination, which is why folding is fast and robust rather than a one-in-a-googol lottery. Partly folded forms, such as the loosely packed molten globule, appear as transient way-stations on the descent. In the cell, much of this happens cotranslationally: the chain begins to fold as it emerges from the ribosome during translation, before synthesis is even complete.
Anfinsen's Dogma: The Sequence Encodes the Structure
In the early 1960s, work on the enzyme ribonuclease produced a foundational result. When the protein was unfolded with denaturing chemicals and its stabilizing disulfide bonds were broken, it lost all activity. When the denaturing conditions were gently removed, the chain spontaneously refolded and recovered its full enzymatic function, with the correct disulfide bonds reforming on their own. No external template or additional information was required.
This led to what is now called Anfinsen's dogma, or the thermodynamic hypothesis: for many small globular proteins, the amino acid sequence alone contains all the information needed to specify the native structure, and that structure corresponds to the conformation with the lowest accessible free energy under physiological conditions. The principle is the conceptual bedrock of the entire field, and it is why predicting structure from sequence is even thinkable. It is not absolute, however. Some proteins fold correctly only with help, depend on cofactors or membrane environments, or remain partly disordered until they bind a partner, which is why folding in a living cell is more complicated than folding in a clean test tube.
Molecular Chaperones: Folding Inside the Crowded Cell
A test tube is dilute and quiet; the cell interior is neither. Protein concentrations inside a cell can reach several hundred grams per liter, and a newly made chain emerges slowly from the ribosome with its sticky hydrophobic regions temporarily exposed. Under these conditions the risk is not just slow folding but irreversible aggregation, where partly folded chains clump together instead of completing their structure. To manage this danger, cells maintain a large set of helper proteins called molecular chaperones.
Chaperones do not contain folding instructions and do not become part of the final product. Instead they bind exposed hydrophobic patches on immature or stress-damaged proteins, shielding them from inappropriate contacts and giving them repeated, protected opportunities to reach the native state. Some, such as the Hsp70 family, act early and engage chains as they are still being synthesized. Others, the chaperonins, form barrel-shaped chambers that enclose a single protein and let it fold in isolation from the crowded cytoplasm. Many chaperones consume ATP to drive cycles of binding and release, linking folding to the cell's energy supply, and the same enzyme machinery is involved in disaggregating and refolding proteins after heat or chemical stress.
This network is one part of a broader system of protein quality control, often called proteostasis, that balances synthesis, folding, and the controlled degradation of proteins that cannot be rescued. The capacity of this system tends to decline with age, which helps explain why many folding-related diseases are age-associated.
Protein Misfolding and Disease
When folding fails, the consequences fall into two broad categories. In loss-of-function disorders, a mutation prevents a protein from reaching its working shape, so the cell is deprived of an activity it needs; cystic fibrosis, in which a common mutation causes the CFTR chloride channel to misfold and be destroyed before it reaches the cell surface, is a classic example. In toxic gain-of-function disorders, the misfolded protein itself becomes harmful, typically by aggregating.
The most studied form of pathological aggregation is the amyloid fibril, a highly ordered, thread-like assembly in which many copies of a protein stack into a stable cross-beta structure. Amyloid and related aggregates are implicated in a striking range of conditions: amyloid-beta and tau in Alzheimer disease, alpha-synuclein in Parkinson disease, huntingtin in Huntington disease, the islet peptide in type II diabetes, and the misfolded prion protein in transmissible spongiform encephalopathies, where a misshapen protein can template the misfolding of its normal counterparts. Smaller, soluble clusters that form on the way to mature fibrils are now thought to be especially toxic to cells.
Because these mechanisms are shared across diseases, folding biology has become a major therapeutic target. Strategies under investigation include small molecules that stabilize a vulnerable protein in its correct shape, treatments that boost the cell's chaperone and clearance capacity, antibodies that recognize and help clear aggregates, and approaches drawn from gene therapy aimed at reducing production of an aggregation-prone protein in the first place. The status of specific drugs and trials in this area changes quickly and should be checked against current clinical sources.
The Protein Folding Problem and AlphaFold
If the sequence specifies the structure, it should be possible, in principle, to compute the folded shape directly from the sequence. Doing so reliably, known as the protein folding problem, remained an unsolved grand challenge for roughly half a century. Determining structures experimentally by X-ray crystallography, nuclear magnetic resonance or cryo-electron microscopy is powerful but slow and expensive, and the number of known sequences vastly outran the number of solved structures, leaving most proteins structurally uncharacterized.
That gap narrowed dramatically with deep learning. In the 2020 community assessment of structure prediction, the AlphaFold system from DeepMind predicted many structures with accuracy approaching that of experiments, a result widely regarded as a turning point for the field. The associated AlphaFold database now provides predicted structures for over 200 million protein sequences, covering nearly all catalogued proteins, and the work was recognized with a share of the 2024 Nobel Prize in Chemistry. These predictions have become a standard starting point across structural biology and bioinformatics.
It is important to be precise about what was solved. AlphaFold predicts many single-chain protein domains with high accuracy, especially well-ordered regions, but confidence drops for intrinsically disordered regions, alternative conformations, ligand-dependent states, membrane-context effects and some complexes. It also does not directly reveal the physical pathway, the order of events, or the timescales by which a chain folds in real time. For many well-ordered proteins, a likely structural endpoint can now be predicted; the kinetics and the mechanisms of misfolding remain active research questions. Understanding those dynamics still matters for designing proteins, interpreting disease mutations, and engineering folding behavior.
Significance in Biotechnology
Folding is a practical constraint, not just a theoretical curiosity. Producing therapeutic proteins through recombinant protein expression frequently fails because overproduced chains misfold and collect into dense, inactive deposits called inclusion bodies; recovering usable product then requires carefully unfolding and refolding the protein. Choosing the right host organism, controlling expression rate, and supplying the right folding environment are central concerns in biotechnology manufacturing, and the folding quality of a biologic drug directly affects its safety, since aggregated protein can provoke immune responses in patients.
Folding knowledge is also generative. Accurate structure prediction and an improving grasp of folding rules now feed into computational protein engineering and the de novo design of proteins that do not exist in nature, including novel enzymes, biosensors and binders. The 2024 Nobel Prize in Chemistry recognized computational protein design alongside structure prediction, underscoring how directly an understanding of folding has translated into the ability to build new molecules to specification rather than only describing existing ones.
Frequently Asked Questions
What determines how a protein folds? For most small proteins, the amino acid sequence alone contains the information needed to reach the correct three-dimensional shape, a principle established by Anfinsen's classic ribonuclease experiments. For many small globular proteins, the native structure is often the most thermodynamically stable accessible state under the relevant conditions. Inside the crowded cell, however, many proteins also rely on molecular chaperones to fold efficiently and to avoid aggregation.
How fast do proteins fold? Small single-domain proteins can fold in microseconds to milliseconds, while larger multidomain proteins may take seconds or longer. This speed is striking given Levinthal's paradox, which notes that randomly sampling every possible conformation would take longer than the age of the universe. Proteins fold quickly because the energy landscape is funnel-shaped and biases the chain toward the native state rather than requiring a random search.
What is the difference between protein folding and protein misfolding? Folding is the productive process by which a polypeptide reaches its functional native structure. Misfolding occurs when the chain settles into a non-native conformation that is non-functional and often prone to aggregation. Misfolded proteins can form amyloid fibrils associated with Alzheimer disease, Parkinson disease, type II diabetes and other disorders.
Does AlphaFold solve the protein folding problem? AlphaFold predicts many single-chain protein domains with high accuracy, especially well-ordered regions, which addresses much of the structure-prediction part of the protein folding problem. Confidence is lower for intrinsically disordered regions, alternative conformations, ligand-dependent states, membrane-context effects and some complexes. It does not by itself reveal the physical pathway or the order of events by which a chain folds in real time. Understanding folding kinetics and misfolding mechanisms remains an active research area.
Can a denatured protein refold? Many small proteins can refold to their active form once the denaturing condition is removed, which demonstrates that the sequence can encode the structure. Larger proteins, proteins with complex topologies, and proteins that depend on chaperones during synthesis often fail to refold efficiently in a test tube and may aggregate instead. Refoldability is therefore protein-specific rather than universal.
Related Terms
Further Reading
Annual Review of Biochemistry, Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade
Signal Transduction and Targeted Therapy, Navigating the Landscape of Protein Folding and Proteostasis: From Molecular Chaperones to Therapeutic Innovations
