RNA Sequencing (RNA-Seq): Methods, Variants, and Clinical Applications
What Is RNA Sequencing (RNA-Seq)?
RNA sequencing (RNA-Seq): A high-throughput transcriptomics method that identifies and quantifies RNA molecules in a biological sample by converting them into sequencing libraries, or in some long-read workflows by reading RNA directly. RNA-Seq shows which genes are active, how strongly they are expressed, which transcript isoforms are present, and how RNA processing changes across cell types, tissues, disease states, or treatments.
RNA-Seq reads the transcriptome: the changing set of RNA molecules produced by a cell, tissue, organoid, tumor, microbe, or mixed biological sample at a particular moment. This makes it different from DNA sequencing, which reads the comparatively stable genome. A genome describes what a cell could do; the transcriptome gives a time- and context-dependent view of what the cell is doing, attempting to do, or responding to.
Most RNA-Seq experiments convert RNA into complementary DNA (cDNA), attach sequencing adapters, and read millions to billions of fragments on a next-generation sequencing platform. The resulting reads are counted, aligned or pseudo-aligned, normalized, and interpreted with bioinformatics tools. Long-read and direct-RNA methods can also sequence longer molecules, sometimes preserving full transcript structures and RNA modification signals that short-read cDNA methods infer only indirectly.
Because RNA abundance is both biologically meaningful and technically fragile, RNA-Seq is best understood as a measurement system rather than a simple readout. The final result depends on sample handling, RNA quality, cell composition, library preparation, sequencing depth, read length, reference annotation, normalization strategy, statistical model, and experimental design. Two well-run RNA-Seq experiments can answer very different questions depending on these choices.
Synonyms and related terms: whole transcriptome shotgun sequencing (WTSS), transcriptome sequencing, transcriptomics sequencing, RNA-seq, expression profiling, single-cell RNA sequencing, spatial transcriptomics, long-read transcriptomics.
Not to be confused with: DNA sequencing, which reads genomic sequence; DNA microarrays, which measure preselected transcripts using probes; or qPCR, which precisely measures one or a small number of RNA targets after reverse transcription.
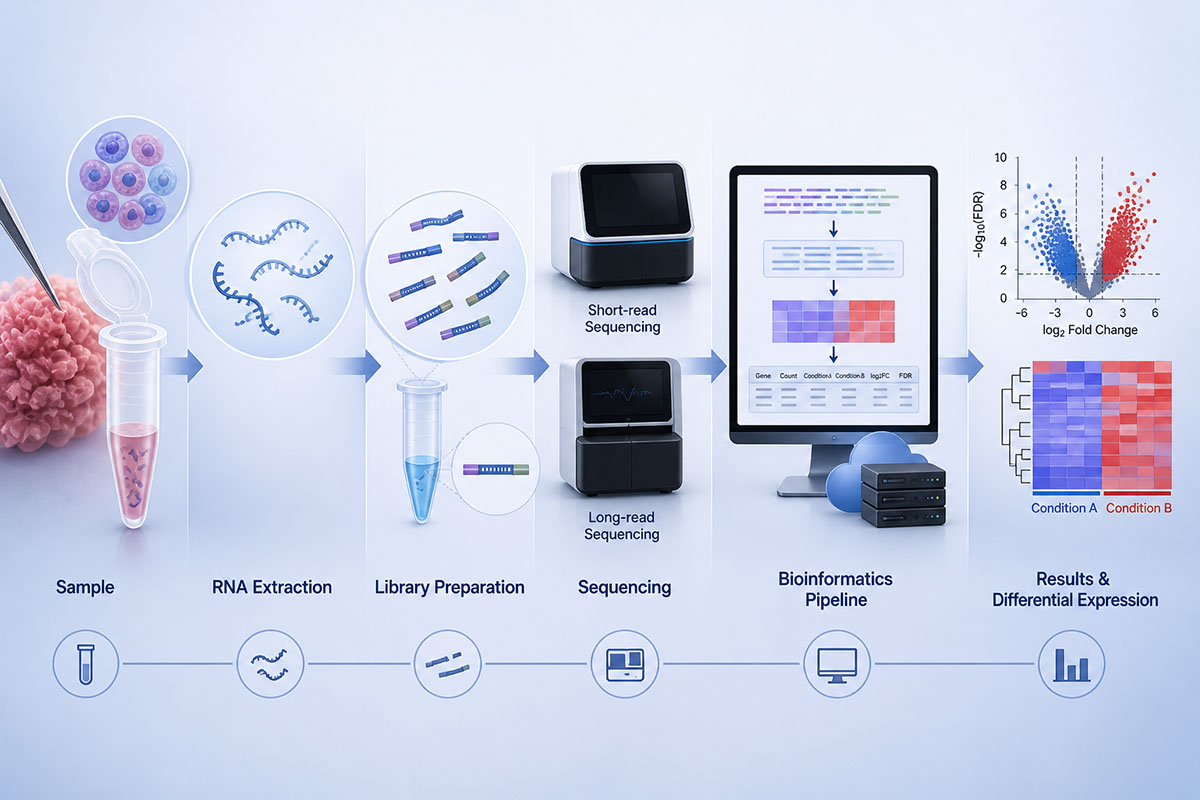
The RNA-Seq workflow: RNA is extracted from a sample, converted to a complementary DNA library or read directly, sequenced on a short-read or long-read platform, and processed through a bioinformatics pipeline that quantifies transcripts and identifies differences between conditions. (Image: Nanowerk) (click on image to enlarge)
Why RNA-Seq Matters
RNA-Seq became a core tool in genomics because it connects genotype, cell state, and phenotype. It can reveal which genes are switched on or off after a drug treatment, which immune cells infiltrate a tumor, which developmental lineage a cell is following, which viral or bacterial transcripts are present in a sample, and which RNA isoforms are produced from the same genome.
Compared with probe-based expression assays, RNA-Seq is open-ended. It can detect previously unannotated transcripts, fusion transcripts, allele-specific expression, splice junctions, long noncoding RNAs, circular RNAs, small RNAs, and pathogen RNA, provided the protocol is designed to capture them. This is why RNA-Seq replaced microarrays for many discovery studies while also becoming a useful companion to targeted assays in clinical and translational research.
The field has also moved beyond bulk tissue averages. Single-cell RNA sequencing separates thousands to millions of cells into individual expression profiles, exposing cell types and transient states that disappear in averaged measurements. Spatial transcriptomics retains tissue position, allowing researchers to ask not only which genes are active, but where those cells and transcripts are located inside a tissue architecture.
How RNA-Seq Works
A typical RNA-Seq experiment has four linked stages: sample collection and RNA isolation, library preparation, sequencing, and computational analysis. The central idea is simple: RNA molecules are sampled, converted into sequenceable molecules, counted, and assigned back to genes or transcripts. The details are what determine whether the experiment measures mature mRNA, total RNA, small RNA, full-length isoforms, single-cell barcodes, spatial coordinates, or native RNA modifications.
1. RNA isolation and quality control
The workflow begins with cells, tissue, blood, organoids, microbes, environmental material, or archived clinical specimens. RNA is chemically unstable and easily degraded by RNases, so collection, freezing, fixation, extraction chemistry, and storage conditions matter. Quality is often assessed with electrophoretic traces and an RNA Integrity Number (RIN), while purity is checked for protein, phenol, salt, or genomic DNA contamination.
For high-quality fresh or frozen mammalian samples, many bulk mRNA workflows use poly(A) selection to enrich messenger RNA. For degraded, microbial, viral, noncoding, or formalin-fixed paraffin-embedded (FFPE) samples, ribosomal RNA depletion is often better because it does not depend on intact poly(A) tails. Choosing between poly(A) selection and rRNA depletion is one of the first major design decisions in RNA-Seq.
2. Library preparation
In most short-read protocols, RNA is reverse-transcribed into cDNA, fragmented or copied from fragmented RNA, ligated to adapters, indexed with sample barcodes, amplified, and size-selected. Strand-specific protocols preserve the direction of transcription, which improves interpretation of overlapping genes, antisense transcripts, and noncoding RNA. Unique molecular identifiers (UMIs) can label original molecules before amplification, helping distinguish true molecule counts from PCR duplicates.
Specialized protocols adapt the same logic to different RNA classes. Small RNA-Seq enriches short regulatory RNAs such as microRNAs. Total RNA-Seq captures coding and noncoding RNA after rRNA depletion. Single-cell protocols add cell barcodes and UMIs to each captured molecule. Spatial protocols add positional barcodes or imaging-based readouts. Long-read protocols aim to preserve full-length transcripts, and direct RNA sequencing can avoid reverse transcription altogether.
3. Sequencing
Most bulk RNA-Seq still uses Illumina short-read sequencing because it is accurate, scalable, and cost-effective for gene-level expression. Typical experiments use single-end or paired-end reads of 50–150 bases, with sequencing depth chosen according to the question: modest depth for abundant gene-expression changes, deeper sequencing for isoforms, rare transcripts, allele-specific expression, or heterogeneous samples.
Long-read sequencing from Pacific Biosciences and Oxford Nanopore Technologies can read full-length transcript molecules or long cDNA molecules. This is especially useful for resolving alternative splicing, transcript start and end sites, fusion transcripts, and complex isoform mixtures. Nanopore sequencing can also read native RNA strands directly, which is valuable for studying RNA modifications and avoiding some amplification biases, although per-read error profiles and throughput considerations differ from short-read workflows.
4. Bioinformatic analysis
Computational analysis turns raw reads into interpretable biological measurements. A bulk workflow usually begins with base-call and adapter quality checks, trimming if needed, alignment to a reference genome or transcriptome, transcript or gene quantification, normalization, exploratory quality control, differential expression testing, and pathway or network interpretation. Common aligners and quantifiers include STAR, HISAT2, Salmon, and kallisto; common differential-expression packages include DESeq2, edgeR, and limma-voom.
For single-cell RNA-Seq, the analysis adds cell calling, barcode correction, UMI counting, doublet detection, filtering, normalization, dimensionality reduction, clustering, marker-gene discovery, cell-type annotation, and trajectory or perturbation analysis. For spatial transcriptomics, computational analysis must also handle tissue images, spots or segmented cells, spatial coordinates, neighborhood structure, and deconvolution when each capture spot contains multiple cells.
Choosing the Right RNA-Seq Strategy
The right RNA-Seq design starts with the biological question, not the instrument or the most fashionable platform. A study asking whether a treatment changes inflammatory pathways in a homogeneous cell line may need simple bulk RNA-Seq. A study asking which rare immune cells respond inside a tumor may need single-cell RNA-Seq. A study asking whether a disease mutation changes transcript isoforms may need long-read RNA-Seq. A study asking where signals occur in tissue may need spatial transcriptomics.
| Question | Best starting approach | Important design choice |
|---|---|---|
| Which genes change between two conditions? | Bulk RNA-Seq | Biological replicates, balanced batches, appropriate normalization |
| Which cell types or states are present? | Single-cell RNA-Seq | Cell viability, dissociation bias, cell number, sequencing depth per cell |
| Which transcript isoforms are produced? | Long-read RNA-Seq or combined short- and long-read analysis | Full-length capture, read accuracy, transcript annotation |
| Where are transcripts located in tissue? | Spatial transcriptomics | Resolution, tissue preservation, imaging, spot or cell segmentation |
| Are small regulatory RNAs changing? | Small RNA-Seq | Size selection, adapter bias, miRNA annotation |
| Are non-polyadenylated RNAs important? | Total RNA-Seq with rRNA depletion | rRNA removal, strand specificity, degraded-sample compatibility |
| Are RNA modifications part of the question? | Direct RNA sequencing or specialized modification assays | Signal-level analysis, negative and positive controls, orthogonal validation |
Sequencing depth is another design variable. More reads do not compensate for poor sample quality, confounded batches, or too few biological replicates. For gene-level differential expression, extra biological replicates and balanced batches are often more valuable than extreme depth. For isoform discovery, fusion detection, single-cell rare-state discovery, or spatial deconvolution, depth and read structure become more important.
Major Variants of RNA-Seq
RNA-Seq is a family of related methods. Each variant captures a different slice of the transcriptome and introduces its own biases. Treating all RNA-Seq data as interchangeable is a common source of overinterpretation.
| Variant | What it measures | Strengths | Main limitations |
|---|---|---|---|
| Bulk RNA-Seq | Average RNA abundance across a mixed sample | Robust, mature, relatively affordable, strong for differential expression | Masks cell-type composition and rare cell states |
| Single-cell RNA-Seq (scRNA-seq) | Expression profiles of individual cells | Reveals heterogeneity, rare populations, trajectories, and cell states | Sparse data, dissociation bias, doublets, shallow coverage per cell |
| Single-nucleus RNA-Seq | RNA from isolated nuclei | Useful for frozen tissue, brain, muscle, and difficult-to-dissociate samples | Enriched for nuclear and pre-mRNA signals; less cytoplasmic RNA |
| Spatial transcriptomics | Expression plus tissue location | Links gene expression to tissue architecture and cell neighborhoods | Resolution, sensitivity, cost, and image-analysis trade-offs |
| Long-read RNA-Seq | Full-length or near-full-length transcripts | Improves isoform, fusion, and transcript-end resolution | Higher cost per base and more complex error handling |
| Direct RNA-Seq | Native RNA molecules | Avoids reverse transcription and can preserve modification-related signals | Lower throughput and specialized analysis requirements |
| Small RNA-Seq | microRNAs, piRNAs, tRNA fragments, and other short RNAs | Targeted view of regulatory small RNAs | Adapter ligation and size-selection biases |
| Total RNA-Seq | Polyadenylated and non-polyadenylated RNA after rRNA depletion | Captures long noncoding RNA, degraded RNA, microbial RNA, and viral RNA | More sequencing spent on abundant non-mRNA species if depletion is incomplete |
Epitranscriptomics has expanded as sequencing technologies have improved. Some RNA modifications can be inferred through specialized enrichment or chemical-conversion methods, while direct RNA sequencing can capture modification-dependent signal changes in native RNA. These methods are powerful, but they require careful controls because modification signals can be confounded by sequence context, structure, coverage, and platform-specific error patterns.
RNA-Seq Compared to Microarrays and qPCR
Before RNA-Seq, researchers studied gene expression mainly with DNA microarrays and qPCR. Microarrays use predesigned probes and are useful when the transcript targets are already known. qPCR measures one or a few transcripts with high sensitivity and remains common for validation, diagnostics, and targeted monitoring. RNA-Seq did not make these older methods obsolete; it changed which method is most appropriate for each question.
| Property | RNA-Seq | DNA microarray | qPCR / RT-qPCR |
|---|---|---|---|
| Discovery potential | High; can detect unannotated transcripts and splice junctions | Limited to probe design | Limited to chosen targets |
| Transcriptome coverage | Whole-transcriptome or targeted, depending on protocol | Predesigned gene or exon probes | One to a few targets per reaction |
| Dynamic range | Broad | Moderate; affected by background and saturation | Very broad for targeted assays |
| Isoform resolution | Good with suitable design; strongest with long reads | Limited | Possible only for preselected isoforms |
| Data complexity | High | Moderate | Low to moderate |
| Best use | Discovery, systems biology, heterogeneity, fusions, isoforms | Known expression panels and legacy datasets | Targeted confirmation and routine assays |
A common practical strategy is to use RNA-Seq for unbiased discovery and qPCR or targeted sequencing for orthogonal confirmation. This is especially important when a finding will be used for mechanistic claims, diagnostic development, or treatment decisions.
How RNA-Seq Data Are Interpreted
RNA-Seq data are often summarized as raw counts, normalized counts, TPM, FPKM, RPKM, log fold change, adjusted p-values, and pathway scores. These quantities are not interchangeable. Raw counts are usually the correct input for differential-expression tools because the statistical model needs count-level information. TPM is useful for comparing relative expression of genes within or across samples in descriptive contexts, but it is not usually the right input for DESeq2 or edgeR differential-expression testing.
Interpretation also depends on the level of quantification. Gene-level counts collapse all isoforms of a gene into one number, which is useful for many expression studies but can hide biologically important transcript switching. Transcript-level quantification can reveal alternative splicing and promoter or polyadenylation changes, but it is more sensitive to read length, annotation quality, and mapping ambiguity. Long reads help resolve this problem by spanning more of each RNA molecule.
Differential expression should not be read as a direct measure of protein abundance or biological causality. mRNA levels can change without corresponding protein changes because translation, protein degradation, localization, feedback loops, and post-translational regulation intervene. Integrating RNA-Seq with proteomics, chromatin accessibility, imaging, and functional assays gives a more complete view of biological state.
Applications in Research and Biotechnology
Differential gene expression and pathway analysis
The most common use of RNA-Seq is to compare transcript abundance between conditions: treated versus untreated cells, diseased versus healthy tissue, resistant versus sensitive tumors, or different stages of development. After statistical testing, researchers often interpret gene sets, pathways, regulators, and networks rather than focusing only on single genes. In gene regulation research, RNA-Seq pairs naturally with chromatin, transcription-factor, and epigenomic assays to connect regulatory inputs to transcriptional outputs.
Cell atlases and developmental biology
Single-cell RNA-Seq has transformed developmental biology, immunology, neuroscience, and organoid research by allowing researchers to reconstruct cell states, lineage trajectories, differentiation programs, and rare populations. Public cell atlases provide reference maps that help annotate new datasets and compare healthy, diseased, treated, and engineered tissues.
Cancer biology and tumor heterogeneity
Bulk RNA-Seq can identify expression subtypes, immune signatures, oncogenic pathway activation, fusion transcripts, and treatment-response programs. Single-cell and spatial approaches add another layer by separating malignant cells from stromal, endothelial, and immune cells and by showing how these populations interact in the tumor microenvironment.
Host-pathogen interactions and infectious disease
RNA-Seq can capture both host responses and pathogen transcripts when sample preparation and reference databases are appropriate. It can distinguish inflammatory states, identify viral RNA, reveal bacterial gene expression programs, and support host-response classifier development, but these studies need strong contamination controls and suitable host, microbial, or viral references. Total RNA-Seq and rRNA-depleted protocols are often important when microbial or viral RNA is part of the question.
Synthetic biology, cell engineering, and perturbation screens
In synthetic biology and cell engineering, RNA-Seq can verify whether engineered circuits, edited cells, induced pluripotent stem cell derivatives, or therapeutic cell products produce the intended transcriptional state. Perturb-seq and related single-cell screens combine genetic perturbations with single-cell RNA-Seq to connect gene function with cellular response programs.
Clinical Applications and Precision Medicine
In oncology, RNA-Seq is valuable because many clinically important events are expressed at the RNA level. It can detect gene fusions, exon skipping, aberrant splicing, expression outliers, immune signatures, and tumor subtype markers. Fusions involving ALK, ROS1, RET, NTRK1, NTRK2, NTRK3, and other genes can guide targeted therapy in selected cancers, and RNA-based assays can sometimes detect expressed rearrangements that DNA-only testing misses.
RNA-Seq can also support rare-disease diagnosis by identifying abnormal splicing, monoallelic expression, expression outliers, or consequences of variants outside coding regions. This can help interpret genomic variants that are difficult to classify from DNA alone. In these settings, tissue choice is critical: the disease-relevant transcript must be expressed in the sampled tissue, or the assay may miss the relevant abnormality.
Single-cell RNA-Seq is generating clinically relevant insights in hematologic malignancies, immunotherapy response, minimal residual disease research, and treatment-resistant cell states. Liquid-biopsy approaches measure circulating mRNA and small RNAs as biomarkers, while host-response transcriptomics can help classify infection or inflammation. These uses are promising, but clinical deployment requires validation, reproducibility, quality control, regulatory review, and clear evidence that the result changes patient management.
RNA-Seq is therefore best viewed as an increasingly important component of personalized medicine, not a standalone answer. It is most powerful when interpreted alongside DNA variants, pathology, imaging, clinical phenotype, treatment history, and orthogonal molecular tests.
Limitations and Common Pitfalls
RNA-Seq is sensitive to both biology and bias. A strong-looking expression difference can reflect a real regulatory change, a shift in cell-type composition, RNA degradation, sample ischemia time, library-preparation bias, sequencing batch, mapping ambiguity, contamination, or a poorly balanced study design. The most important safeguard is experimental design: enough biological replicates, randomized processing, balanced batches, predefined contrasts, and metadata that capture age, sex, tissue source, treatment timing, quality metrics, and other covariates.
| Pitfall | Why it matters | Actionable safeguard |
|---|---|---|
| RNA degradation | Creates 3′ bias and loss of full-length information | Control collection time, storage, extraction, RIN or DV200, and protocol choice |
| Batch effects | Can dominate true biological differences | Randomize samples, balance groups across batches, include covariates, inspect PCA/UMAP plots, and avoid confounding condition with run date |
| Too few replicates | Weakens statistical power and inflates false discoveries | Prioritize biological replication over unnecessary sequencing depth |
| Cell-type composition changes | Bulk differences may reflect different cell mixtures | Use deconvolution, sorted cells, single-cell RNA-Seq, or spatial transcriptomics |
| Mapping ambiguity | Paralogs, repeats, pseudogenes, and isoforms can confuse quantification | Use appropriate references, transcript-aware tools, and validation for key findings |
| Overinterpreting fold change | Large fold changes in low-count genes may be unstable | Inspect counts, dispersion, adjusted p-values, effect sizes, and biological plausibility |
| Single-cell dropout, doublets, and ambient RNA | Create false absence, mixed-cell profiles, or misleading background expression | Use UMI-aware pipelines, doublet and ambient-RNA correction, filtering, and suitable validation |
| Annotation bias | Known transcripts are easier to detect than novel ones | Use updated annotations, long reads, and discovery-aware tools when isoforms matter |
| Reference and contamination errors | Misassigned reads can create false pathogen, fusion, or paralog signals | Use appropriate reference genomes, spike-ins or controls where useful, and orthogonal confirmation for consequential findings |
Single-cell and spatial datasets add specific interpretation risks. Cell dissociation can selectively lose fragile or adherent cell types. Stress-response genes may turn on during handling. Cell-type labels can be subjective when marker genes overlap. Spatial spots may contain multiple cells, and image segmentation errors can propagate into expression analysis. These are not reasons to avoid the methods; they are reasons to design controls and interpret results at the resolution the data actually support.
Typical Research Workflow
A robust RNA-Seq project usually follows a staged workflow rather than jumping directly from sequencing to biological claims.
- Define the biological question. Decide whether the study needs gene-level expression, isoforms, cell types, tissue location, pathogen RNA, small RNAs, or RNA modifications.
- Choose the protocol. Select bulk, single-cell, single-nucleus, spatial, small RNA, total RNA, long-read, or direct RNA sequencing based on the question and sample type.
- Design the experiment. Plan biological replicates, controls, randomization, batch balancing, sample metadata, and quality thresholds before sequencing begins.
- Prepare and sequence libraries. Track extraction, RNA quality, library size, strandedness, depth, barcodes, and platform-specific quality metrics.
- Run primary analysis. Perform read quality checks, alignment or quantification, count generation, filtering, normalization, and exploratory analysis.
- Test hypotheses. Use suitable statistical models for differential expression, isoform usage, cell-state differences, spatial neighborhoods, or perturbation effects.
- Interpret biology carefully. Examine effect sizes, genes, pathways, cell types, and possible confounders rather than relying on a single ranked gene list.
- Validate key findings. Confirm important results with qPCR, targeted sequencing, protein assays, imaging, functional experiments, or independent cohorts when conclusions require high confidence.
Key Terms Related to RNA-Seq
Read count: The number of sequencing reads assigned to a gene, transcript, exon, splice junction, barcode, or feature.
TPM: Transcripts per million, a length- and library-size-normalized expression measure often used for descriptive comparisons.
FPKM/RPKM: Older length- and depth-normalized measures. They still appear in legacy datasets but are often less preferred than TPM for descriptive expression comparisons.
UMI: Unique molecular identifier, a short random barcode attached to an original RNA or cDNA molecule to help count molecules and remove PCR duplicates.
Strandedness: Information about which DNA strand corresponds to the original RNA transcript. Stranded libraries improve interpretation of antisense and overlapping transcripts.
Splice junction: A sequence read spanning two exons that were joined during RNA splicing. Junction reads are central to isoform and fusion analysis.
Batch effect: A technical difference between groups of samples, such as extraction date, sequencing run, technician, kit lot, or instrument, that can mimic biological differences.
Dropout: In single-cell RNA-Seq, the apparent absence of an RNA molecule that was biologically present but not captured or sequenced.
Differential expression: A statistical comparison of RNA abundance between conditions, usually reported with log fold changes and multiple-testing-adjusted significance values.
Future Directions
RNA-Seq is moving toward richer measurements rather than simply more reads. Direct RNA sequencing is improving access to native RNA molecules and modification-aware analysis. Long-read methods are becoming more practical for isoform-resolved transcriptomics. Single-cell methods are integrating RNA with chromatin accessibility, surface proteins, CRISPR perturbations, lineage recording, and spatial information. Spatial methods are moving toward finer resolution, larger tissue areas, and three-dimensional tissue reconstruction.
Machine-learning models trained on large expression atlases are beginning to support cell-type annotation, perturbation prediction, quality control, batch integration, and cross-dataset search. These models can accelerate analysis, but they also import the biases of the datasets used to train them. The most useful future RNA-Seq studies will combine high-quality experimental design with transparent computational methods and validation, rather than relying on sequencing scale alone.
Frequently Asked Questions
How long does an RNA-Seq experiment take from sample to results? A typical non-clinical bulk experiment often takes days to a few weeks from RNA extraction to an initial analysis, depending on sequencing queue, library-preparation workflow, and computational capacity. Single-cell, spatial, long-read, FFPE, low-input, or clinical-grade projects often take longer because sample handling, quality control, data processing, and interpretation are more complex.
What units are used to report RNA-Seq results? Common units include raw counts, normalized counts, TPM, FPKM/RPKM, counts per million, log fold change, and adjusted p-values. Raw counts are usually used for statistical differential-expression tools, while TPM is often used for descriptive expression comparisons.
How much input RNA is needed for RNA-Seq? Standard bulk protocols often use tens of nanograms to microgram-scale total RNA, while low-input protocols can work with much less. Single-cell RNA-Seq starts from the RNA content of individual cells or nuclei and therefore relies on barcoding and amplification. The practical input requirement depends on sample quality, protocol, organism, and whether the goal is gene expression, isoforms, small RNA, or degraded clinical material.
Can RNA-Seq be performed on FFPE tissue samples? Yes, but FFPE RNA is usually fragmented and chemically modified. FFPE-compatible workflows often use random priming, rRNA depletion, short inserts, or targeted panels. Results can be highly useful for retrospective clinical research, but the protocol should be chosen for degraded RNA and interpreted with quality metrics such as DV200.
Is RNA-Seq quantitative? RNA-Seq is quantitative within limits. It provides digital counts related to molecule abundance, but counts are affected by gene length, library size, composition bias, capture efficiency, GC content, mapping ambiguity, transcript annotation, and sample quality. Strong claims should use suitable normalization, statistical modeling, replicates, and validation.
What software tools are commonly used for RNA-Seq analysis? Standard bulk pipelines commonly use FastQC or MultiQC for quality checks, STAR or HISAT2 for alignment, Salmon or kallisto for transcript quantification, featureCounts or HTSeq for gene counts, and DESeq2, edgeR, or limma-voom for differential expression. Single-cell analysis commonly uses Seurat, Scanpy, scVI, Cell Ranger, and related ecosystem tools, while workflow managers such as Nextflow and Snakemake help make pipelines reproducible.
When should long-read RNA-Seq be used instead of short-read RNA-Seq? Long-read RNA-Seq is most useful when full-length transcript structure matters: alternative splicing, fusion transcripts, allele-specific isoforms, transcript start and end sites, or complex gene models. Short reads remain efficient for many gene-level expression studies, and combined short- and long-read designs are often stronger than either alone.
Selected References
Nature Reviews Genetics, RNA Sequencing: The Teenage Years
Nature Reviews Genetics, Best Practices for Single-Cell Analysis Across Modalities
Nature Reviews Genetics, Methods and Applications for Single-Cell and Spatial Multi-Omics
Nature Reviews Clinical Oncology, Emerging Clinical Applications of Single-Cell RNA Sequencing in Oncology
Nature Methods, A Systematic Benchmark of Nanopore Long-Read RNA Sequencing for Transcript-Level Analysis in Human Cell Lines
Nature Portfolio / npj Systems Biology and Applications, A Benchmark of RNA-seq Data Normalization Methods
