| Posted: Feb 11, 2015 |
Molecular dynamics unravel the dependence of friction on the atomistic details of surfaces |
| (Nanowerk News) Researchers at UCL in a collaboration study with Laurent Joly, from the University of Lyon, have investigated the friction properties of liquid water at the interface with graphene and with an hexagonal boron nitride sheet, using ab initio molecular dynamics for the first time (Nature Nanotechnology, "Simulating water slippage"). |
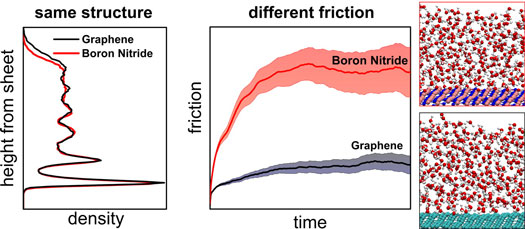 |
| ab initio molecular dynamics can be used to unravel the connection between the structure of nanoscale water and friction for liquid water in contact with graphene and with hexagonal boron nitride. Although the interface presents a very similar structure between the two sheets, the friction coefficient on boron nitride is ∼3 times larger than that on graphene. This comes about because of the greater corrugation of the energy landscape on boron nitride arising from specific electronic structure effects. |
| Friction is one of the main sources of dissipation and is the main factor limiting the efficiency of nanofluidic devices. For instance, about one third of the world's mechanical energy is dissipated into friction, and so an understanding of nanoscale friction at the interface between a liquid and a solid is also crucial for the development of efficient membranes for water desalination and power harvesting. |
| Although previous work observed a correlation between friction and wetting, clear understanding of its relation to structure and energetics of the liquid/solid interface remains elusive. Such understanding is of paramount importance for the prediction of the friction properties of functional surfaces and for the rational design of materials with optimal friction. |
| Boron nitride (BN) is attracting increasing attention in nanofluidic applications, mainly because of potential applications of BN nanotubes in e.g. blue energy harvesting or water filtration. BN is also thought to have similar wetting properties to carbonaceous materials, but whether BN nanostructures share the superlubricating properties of carbon nanotubes remains unclear. |
| This UCL-led collaborative study showed for the first time that liquid/solid friction can be accurately quantified using ab initio molecular dynamics. This is an achievement that, mainly due to massive advances in computational power, would have been inconceivable just a few years ago. With the ab initio approach used, the friction properties of water on carbon and BN nanostructures were compared on a completely equal footing. This revealed the quite surprising result that whilst the structure and wetting properties of the two interfaces are extremely similar, friction is very different. |
| This work makes a crucial step toward understanding and predicting liquid/solid friction in nanofluidic systems, and is envisaged to have a strong impact on the community. In addition the demonstration that ab initio methods can be used to determine the frictional properties of interfaces opens up new avenues of research for the nanofluidic community, such as the ability to realistically explore the role of defects or charge transfer at interfaces. |
| Source: London Centre for Nanotechnology |

