| Posted: Oct 04, 2017 |
The road less traveled: How to switch assembly pathways(Nanowerk News) Understanding how the structure of the constituent molecules controls materials assembly could advance materials design. However, mapping out early formation steps is difficult. Now, these first steps have been uncovered in real time for 2-D crystalline sheets and 3-D porous networks from biologically inspired polymers. The sequence in which the individual molecules within the polymers linked up determined the assembly pathway: a direct, one-step process or a more complex, two-step path. Tuning the structure of the starting molecules controlled assembly. |
| The study (Nature Materials, "Tuning crystallization pathways through sequence-engineering of biomimetic polymers") offers new rules for designing materials. The rules could permit energy-efficient synthesis of materials with properties not offered today. Using these rules, scientists could build materials for manufacturing that, for example, lead to better solar panels. The work also resolves a long-standing controversy over crystal growth. |
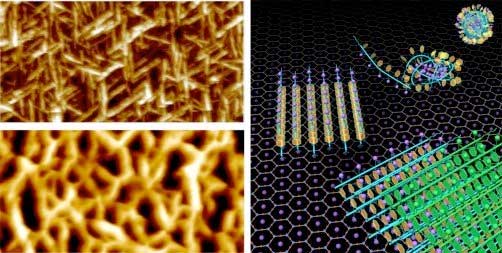 |
| On the left, atomic force microscopy images show the structures formed from a polymer with a hydrophobic (water-fearing) tail via a two-step nucleation and growth process. The top figure shows the crystal during the assembly process. The bottom figure shows the final self-assembled network. On the right, the two-step formation process is depicted. Starting from the top right corner and going counterclockwise, the clusters in solution deposit onto the surface, transform into single molecule-wide linear structures and spread laterally as new linear structures nucleate on top to form vertical 2-D crystals. (Image: Pacific Northwest National Laboratory) |
| Bioinspired assembly of complex building blocks that vary in structure and properties form ordered 2-D and 3-D structures. These assembly pathways are being actively pursued to create materials with new properties not achievable using existing synthetic methods. However, little information about how these dynamic processes occur at the fundamental level is available. |
| Scientists at Pacific Northwest National Laboratory and Lawrence Livermore National Laboratory exploited the sub-nanometer resolution of atomic force microscopy and the tendency of surfaces to promote crystallization to observe in real time assembly pathways for biomimetic peptoid polymers. Two identical polymers were synthesized, but one had a hydrophobic (water-fearing) tail linked to its end. The polymers were added to solutions containing calcium ions and formed 2-D crystalline sheets and 3-D porous networks on surfaces, which was observed in real time. |
| Final assembly of the porous networks was preceded by the formation of arrays of 5-nanometer-wide, 3-nanometer-high linear structures that grew to form vertically oriented sheets aligned along three directions. The polymer without a tail followed a one-step process as expected with monomers readily forming crystalline particles in solution and depositing on the surface at a rate that decreases as coverage increases. |
| Adding the short hydrophobic “tail” caused nucleation to take a two-step path. The first step was the aggregation of 10 to 20 polymers into disordered clusters deposited on the surface. In the second step, the clusters suddenly rearranged themselves to “stand up” into crystalline particles with heights of 3 nanometers that then grew — as one molecule after another snapped into place. Conversely, a polymer with a hydrophilic (water-loving) rather than hydrophobic tail followed the direct, one-step process. |
| This demonstrates that creation of the disordered clusters, whose formation and transformation controls the rate of crystallization, arises from the increased propensity for collective polymer aggregation as a result of the hydrophobicity of the linked tails. |
| The results showed that the pathways are indeed dependent on the polymer sequence of each building block and have significant implications for the design of self-assembling polymer systems. |
| Source: U.S. Department of Energy, Office of Science |

