| May 04, 2022 |
New AI tool models in record time the behavior of clusters of nanoparticles
(Nanowerk News) New AI tool models in record time the behavior of clusters of nanoparticles ("Learning in continuous action space for developing high dimensional potential energy models").
|
|
We learn new skills by repetition and reinforcement learning. Through trial and error, we repeat actions leading to good outcomes, try to avoid bad outcomes and seek to improve those in between. Researchers are now designing algorithms based on a form of artificial intelligence that uses reinforcement learning. They are applying them to automate chemical synthesis, drug discovery and even play games like chess and Go.
|
|
Scientists at the U.S. Department of Energy’s (DOE) Argonne National Laboratory have developed a reinforcement learning algorithm for yet another application. It is for modeling the properties of materials at the atomic and molecular scale and should greatly speed up materials discovery. |
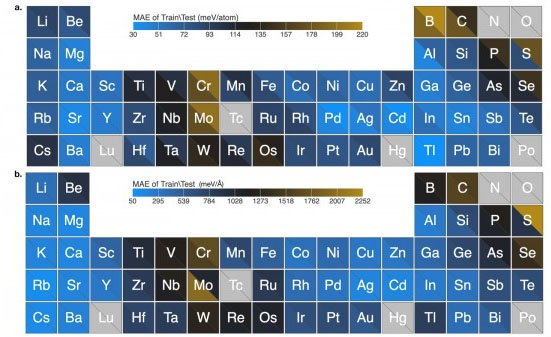 |
| Graph shows excellent algorithm performance for force field predictions of elemental nanoclusters covering 54 elements in the periodic table. Data demonstrate very low mean absolute error. (Image: Argonne National Laboratory)
|
|
Like humans, this algorithm “learns” problem solving from its mistakes and successes. But it does so without human intervention.
|
|
Historically, Argonne has been a world leader in molecular modeling. This has involved calculating the forces between atoms in a material and using that data to simulate its behavior under different conditions over time.
|
|
Past such models, however, have relied heavily on human intuition and expertise and have often required years of painstaking efforts. The team’s reinforcement learning algorithm reduces the time to days and hours. It also yields higher quality data than possible with conventional methods.
|
|
“Our inspiration was AlphaGo,” said Sukriti Manna, a research assistant in Argonne’s Center for Nanoscale Materials (CNM), a DOE Office of Science user facility. “It is the first computer program to defeat a world champion Go player.”
|
|
The standard Go board has 361 positional squares, much larger than the 64 on a chess board. That translates into a vast number of possible board configurations. Key to AlphaGo becoming a world champion was its ability to improve its skills through reinforcement learning.
|
|
The automation of molecular modeling is, of course, much different from a Go computer program. “One of the challenges we faced is similar to developing the algorithm required for self-driving cars,” said Subramanian Sankaranarayanan, group leader at Argonne’s CNM and associate professor at the University of Illinois Chicago.
|
|
Whereas the Go board is static, traffic environments continuously change. The self-driving car has to interact with other cars, varying routes, traffic signs, pedestrians, intersections and so on. The parameters related to decision making constantly change over time.
|
|
Solving difficult real-world problems in materials discovery and design similarly involves continuous decision making in searching for optimal solutions. Built into the team’s algorithm are decision trees that dole out positive reinforcement based on the degree of success in optimizing model parameters. The outcome is a model that can accurately calculate material properties and their changes over time.
|
|
The team successfully tested their algorithm with 54 elements in the periodic table. Their algorithm learned how to calculate force fields of thousands of nanosized clusters for each element and made the calculations in record time. These nanoclusters are known for their complex chemistry and the difficulty that traditional methods have in modeling them accurately.
|
|
“This is something akin to completing the calculations for several Ph.D. theses in a matter of days each, instead of years,” said Rohit Batra, a CNM expert on data-driven and machine learning tools. The team did these calculations not only for nanoclusters of a single element, but also alloys of two elements.
|
|
“Our work represents a major step forward in this sort of model development for materials science,” said Sankaranarayanan. “The quality of our calculations for the 54 elements with the algorithm is much higher than the state of the art.”
|
|
Executing the team’s algorithm required computations with big data sets on high performance computers. To that end, the team called upon the carbon cluster of computers in CNM and the Theta supercomputer at the Argonne Leadership Computing Facility, a DOE Office of Science user facility. They also drew upon computing resources at the National Energy Research Scientific Computing Center, a DOE Office of Science user facility at Lawrence Berkeley National Laboratory.
|
|
“The algorithm should greatly speed up the time needed to tackle grand challenges in many areas of materials science,” said Troy Loeffler, a computational and theoretical chemist in CNM. Examples include materials for electronic devices, catalysts for industrial processes and battery components.
|

