| Posted: Feb 08, 2015 |
'Virtual virus' unfolds the flu on a CPU
|
|
(Nanowerk News) By combining experimental data from X-ray crystallography, NMR spectroscopy, cryoelectron microscopy and lipidomics (the study of cellular lipid networks), researchers at the University of Oxford have built a complete model of the outer envelope of an influenza A virion for the first time. The approach, known as a coarse-grained molecular dynamics simulation, has allowed them to generate trajectories at different temperatures and lipid compositions - revealing various characteristics about the membrane components that may help scientists better understand how the virus survives in the wild or find new ways to combat it.
|
|
Their computer simulation begins by rendering the virus as a relatively large, 73-nanometer ball of loosely packed lipids. This ball then relaxes down into a smaller, 59-nanometer virion within 300 nanoseconds - an imperceptible amount of time on the macroscopic level, but roughly 1/15th of the simulation's total run time. The viral spike proteins are then embedded into the lipid envelope individually, before adding solvent to the system.
|
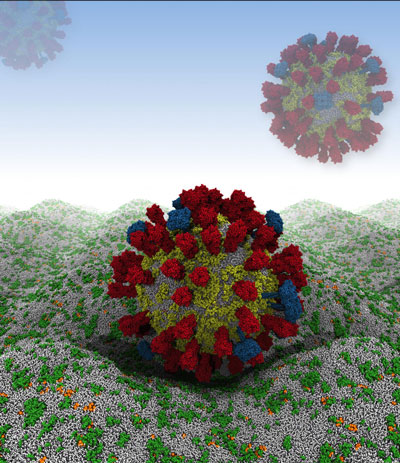 |
| Future simulation work will involve the influenza A virus in close apposition with a host cell membrane. (Image: H. Koldso/Oxford)
|
|
"In the current publication, this is just a single virion in a water droplet -- but what could really get interesting is if we start putting in other molecules of interest, such as therapeutic agents," said Tyler Reddy, a postdoctoral fellow at the University of Oxford. Reddy will describe the group's findings this week at the Biophysical Society's 59th annual meeting in Baltimore, Md.
|
|
From the simulation, Reddy and coworkers have found that the viral spike proteins protruding from the virion's membrane spread out, rather than aggregating close together. This is key to the strength of the interactions between influenza A virions and host cells, which are determined by the number of spike proteins that can engage with receptors.
|
|
"If the separation of the spike proteins is compatible with the 'arms' of Y-shaped, bivalent antibodies, this information might be exploited in therapeutic design, so that two antigens may be bound simultaneously for enhanced association," Reddy said.
|
|
Reddy's work builds on that of Daniel Parton, whom Reddy met while visiting Oxford in 2010. Parton is a co-author on the paper Reddy's talk is based on and currently a postdoctoral fellow at the Memorial Sloan Kettering cancer Center in New York City. Reddy, at the time, was working with molecular dynamics simulations for a membrane protein system relevant to achondroplasia, the most common cause of human dwarfism. He was impressed by Parton's plan to construct and simulate the lipid envelope of the entire flu virion, and later decided to take aim at it himself when applying for a postdoctoral fellowship at Oxford.
|
|
The researchers also observed that the Forssman glycolipid had a role in preventing protein aggregation and slowing down protein diffusion. Reddy indicated it would be important to include glycolipids in future virion simulations given their influence on the biophysical properties observed. The extended sugar head groups of glycolipids may also mask antibody accessibility of the M2 proton channels in the flu envelope -- the target of commonly prescribed anti-influenza drugs based on adamantane derivatives.
|
|
Understanding the membrane envelope's structural dynamics also provides insight into the wide-ranging survival times of the virion in different environments, such as fresh-water rivers. Previous studies have indicated that the presence of influenza A in rivers has allowed waterfowl to be simultaneously exposed to source flu strains and residual anti-viral compounds excreted by local human populations, potentially giving rise to drug-resistant influenza strains. Reddy's simulation currently monitors the virion's stability on the micro-second scale, and it will be a challenge to assess stability over much longer time scales.
|
|
"We are a long way from being able to perform molecular dynamics simulations that span the year time scale," Reddy said. "Nonetheless, we now have a platform for looking at influenza A virion behavior in silico, and perhaps certain compounds or solutions could be used to accelerate destabilization on an observable time scale," Reddy said.
|
|
"We're making the coordinate data freely available in the hopes that other groups have interesting ideas for use with this model as well, and so that they can criticize and help improve the model."
|
|
The presentation, "Nothing to sneeze at: a full-scale computational model of the human influenza virion" by Tyler Reddy, David Shorthouse, Daniel Parton, Elizabeth Jefferys, Philip W. Fowler, Matthieu Chavent, Marc Baaden and Mark S.P. Sansom is at 10:45 AM, on Sunday, Feb. 8, 2015 at the Baltimore Convention Center, room 316/317.
|