| Aug 21, 2012 |
A team of scientists move toward rational design of artificial proteins
|
|
(Nanowerk News) In the world of proteins, form defines function. Based on interactions between their constituent amino acids, proteins form specific conformations, folding and twisting into distinct, chemically directed shapes. The resulting structure dictates the proteins’ actions; thus accurate modeling of structure is vital to understanding functionality.
|
|
Peptoids, the synthetic cousins of proteins, follow similar design rules. Less vulnerable to chemical or metabolic breakdown than proteins, peptoids are promising for diagnostics, pharmaceuticals, and as a platform to build bioinspired nanomaterials, as scientists can build and manipulate peptoids with great precision. But to design peptoids for a specific function, scientists need to first untangle the complex relationship between a peptoid’s composition and its function-defining folded structure.
|
|
Past efforts to predict protein structure have met with limited success, but now a scientific team led by Glenn Butterfoss, and Barney Yoo, research scientists at New York University, in collaboration with investigators from the U.S. Department of Energy’s Lawrence Berkeley National Laboratory (Berkeley Lab), Stony Brook University and Temple University have demonstrated that a computer modeling approach similar to one used to predict protein structures can accurately predict peptoid conformation as well.
|
|
The authors describe this accomplishment in a new paper in the Proceedings of the National Academy of Sciences ("De novo structure prediction and experimental characterization of folded peptoid oligomers").
|
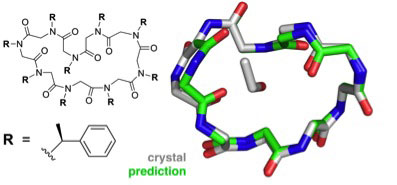 |
| Predicted structure of the cyclic nonamer proposed by the theorists, shown to match the actual folded structure with remarkable accuracy.
|
|
“Natural selection has engineered protein sequences that can self-assemble into molecular machines with specific functions. Why can’t we do the same with biologically inspired synthetic materials?” Voelz, Principal Investigator with Temple University, explains.
|
|
With this mission in mind, the collaborative team of scientists developed the project two years ago at the 7th Peptoid Summit, a – conference devoted to peptoid research hosted by Berkeley Lab’s Molecular Foundry.
|
|
“The research was carried out by a remarkable, interdisciplinary team of scientists,” says Kent Kirshenbaum of NYU. “Some of the team have worked together on this truly difficult problem for almost 20 years. The researchers include both experimentalists and theorists who have been able to guide one another in discovering how these peptoid molecules fold.”
|
|
Together, they proposed a ‘blind structure prediction’ challenge. This self-assessment technique, responsible for the enormous progress in the world of protein structure modeling, allows scientists to test the fidelity of their computational models by predicting the three-dimensional structure of a known molecule and then comparing their proposed structure to the X-ray crystallography results.
|
|
An analogous, combined experimental-computational method was employed by the peptoid team in an effort to advance the computational design of peptoid structure. X-ray crystal structures for three peptoid molecules, two small and linear and one larger and cyclical, were simultaneously determined, but not disclosed to the theoretical modelers. The experimentalists then used a combination of two simulation techniques, Replica Exchange Molecular Dynamics (REMD) simulation and Quantum Mechanical refinement (QM). REMD can efficiently predict the preferred general conformations, and the QM calculations further refine the conformational prediction. In combination, these two calculations accurately define the physical structures of molecules.
|
|
The proposed structural predictions of the peptoid molecules did exceedingly well at calculating the actual folded conformations. The first two blind predictions were calculated for two linear, small N-alkyl and N-aryl peptoid trimers. Of these, the N-aryl peptoid trimer was the best blind prediction, matching the crystal described conformation to within 0.2 Å. The N-alkyl trimer prediction matched less well with the crystal results because of its increased flexibility.
|
|
The greater challenge facing the group was structural prediction of the larger, cyclic peptoid nonamer. Six different possible conformations were considered for the final, submitted prediction and the top pick proved to agree best with the crystallography results to an accuracy of 1.0 Å.
|
|
This success suggests that reliable structure prediction for complex three-dimensional folds is within reach, an enormous step forward on the path to reliable and efficient computational peptoid design.
|
|
“This will hopefully break open the field of peptoid structure prediction and design, an area we desperately need to guide our more well-developed synthetic efforts,” says Ron Zuckermann, co-author and director of the Biological Nanostructures Facility at Berkeley Lab’s Molecular Foundry.
|
|
“It is an exciting time for peptoid research,” says author Glenn Butterfoss, research scientist with NYU. “The community of labs working on these molecules is growing, and both the diversity and creativity of recent studies is quite astonishing. We hope our work here, aimed toward understanding the structural behavior of peptoids in three dimensional space, serves as a building block for future efforts to design peptoid molecules with practical functions.”
|
|
Portions of this work were performed at the Molecular Foundry, Lawrence Berkeley National Laboratory, and supported by the DOE Office of Science. Additional funding came from the Defense Threat Reduction Agency, the NSF and the NIH.
|