| Aug 29, 2017 |
New open-source software for analyzing intact proteins
|
|
(Nanowerk News) An estimated 20,300 genes in the human genome encode proteins. The number of proteins themselves, as intact proteoforms, could be as high as one billion.
|
|
That vast number makes the functional protein architecture of humans - called the proteome - much harder to characterize than the genome.
|
|
Yet characterizing the proteome is essential for understanding the activities and functions of proteins that mediate the diagnosis, treatment, and prevention of disease. It is also necessary for understanding proteins that exist in environments outside the human body.
|
|
Typically, the proteomics data used to characterize the proteome are collected by liquid chromatography-mass spectrometry (LC-MS) analytical strategies. Instrumentation like this is designed to reveal the function and activity of proteins by accurately measuring charge, mass, and weight.
|
|
A new Nature Methods paper ("Informed-Proteomics: open-source software package for top-down proteomics") by lead author Jungkap Park and fellow scientists at the Pacific Northwest National Laboratory (PNNL) introduces Informed-Proteomics, a novel open-source suite of software for identifying intact proteins from mass spectrometry analysis. It contains a full suite of novel software tools for top-down proteomics, which is used to analyze intact proteins.
|
|
Efficient and streamlined, Informed-Proteomics offers substantial improvements over current methods by offering a new LC-MS feature-finding algorithm, a new database search algorithm, semi-automated learning methods, and an interactive results viewer.
|
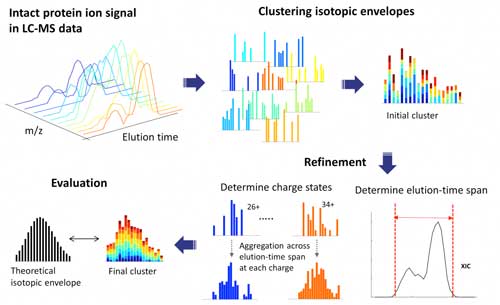 |
| ProMex LC-MS Feature Finding: The algorithm begins with clustering isotopomer envelopes across adjacent time and charge state. The initial cluster is refined to accurately determine its elution-time span and range of charge states. After refinement, ProMex calculates the likelihood that the final cluster is an LC-MS feature. (click on image to enlarge)
|
Studying a Protein's ‘Native Structure'
|
|
In the traditional "bottom-up" proteomics methodology, proteins are digested into peptides for mass spectrometry identification. This method offers higher throughput, but the results can be inconclusive regarding the intact and active protein form.
|
|
The top-down method analyzes each protein while the molecule is intact. In this way, top-down proteomics preserves valuable information about post-translational modifications, isoforms, and the molecular combinations that are collectively called proteoforms.
|
|
"Studying a protein in its native structure is important" since so much more information about the protein is preserved, said co-author Sam Payne, a PNNL Integrative Omics scientist and team lead. However, he added, "there are very unique challenges to studying the protein as a whole."
|
|
Among the technical hurdles of top-down proteomics is "getting to the scale you want to be," said Payne. The spectra derived from top-down methods are much more complex, and require new software tools and novel algorithms to meet what he called the "hugely challenging" idea of measuring all the proteins in a cell.
|
|
"With top-down, what you look for is extraordinarily large," said Payne - and that requires the right mathematics "to organize an efficient way to search."
|
‘Search Space,' and a Breast Cancer Test
|
|
Why so large a scale? For one, in top-down proteomics the size of intact proteins means the signal after ionization is spread out over many dimensions. For another, what Payne called the "search space" of potential proteoforms is very big. The combinatory universe of proteins can number up to a billion.
|
|
The authors evaluated Informed-Proteomics alongside several other popular top-down proteomics tools by using human-in-mouse xenograft luminal and basal breast tumor samples that are known to have significant differences.
|
|
In analyzing over 3,000 proteoforms in two breast cancer subtypes, the PNNL authors saw that their new software tool found ten times more differentially expressed proteoforms compared to a recent top-down analysis using a different method.
|
|
One advantage for the PNNL authors comes from PNNL's "very long history in leading top-down analysis" in both instruments and informatics, said Payne, a fact that reflects the work of co-author Richard D. Smith. "As a team, we can make improvements in all aspects of the analysis, both computational and technological."
|
|
Currently, the quality of datasets from liquid chromatography and mass spectrometry instrumentation is universally increasing, along with the quality of sample-processing protocols. With substantially more complex top-down mass spectra to deal with, the paper's authors report "an urgent need to develop algorithms and software tools for confident proteoform identification and quantification."
|