| Aug 04, 2021 |
New tool enables mapping of protein interaction networks at scale
|
|
(Nanowerk News) Proteins—long chains of amino acids—each play a unique role in keeping our cells and bodies functioning, from carrying out chemical reactions, to delivering messages, and protecting us from potentially harmful foreign invaders. More recent research has shown that these proteins not only serve their individual purpose, but also interact with other proteins to carry out even more numerous and complex functions through these protein-protein interactions (PPI).
|
|
Collectively, all the protein-protein interactions in a cell form a PPI network. Experimentally identifying a PPI network within human cells has required a tremendous amount of time and resources, with experiments required to identify every individual PPI, and many additional experiments to investigate these protein pairs for network-level interactions.
|
|
Now, bioengineers at the University of California San Diego have developed a technology capable of revealing the PPIs among thousands of proteins, in a single experiment. The tool, called PROPER-seq (protein-protein interaction sequencing), allows researchers to map the PPI network from their cells of interest within several weeks, without any specialized resources such as antibodies or premade gene libraries.
|
|
The researchers describe this technology in Molecular Cell ("Revealing protein-protein interactions at the transcriptome scale by sequencing"). They applied PROPER-seq on human embryonic kidney cells, T lymphocytes, and endothelial cells, and identified 210,518 PPIs involving 8,635 proteins.
|
 |
| Visualization of a protein-protein interaction network identified using PROPER-seq. (Image: UCSD)
|
|
"PROPER-seq is capable of scanning the order of 10,000×10,000 protein pairs in one experiment," said Kara Johnson, a recent UC San Diego bioengineering Ph.D. alumna and the first author of this paper. The research was conducted in bioengineering professor Sheng Zhong’s lab.
|
|
The central idea of PROPER-seq is to label every PPI with a unique DNA sequence, and then read these DNA sequence labels through next-generation sequencing. To implement this idea, Zhong's team developed a technique called SMART-display, which attaches a unique DNA barcode to every protein. They also devised a method called "Incubation, ligation and sequencing" (INLISE) to sequence the pair of DNA barcodes that are attached to two interacting proteins. The third component of PROPER-seq is a software package called PROPERseqTools, that incorporates statistical tools to identify the PPIs from the DNA sequencing data. This trio of tools—SMART-display, INLISE, and PROPERseqTools—together is known as PROPER-seq.
|
|
A laboratory would start the PROPER-seq protocol with their cells of interest, and obtain the output as a list of identified PPIs. The user can also get the DNA sequence read counts and other statistics associated with every identified PPI.
|
|
Zhong's team applied PROPER-seq on human embryonic kidney cells, T lymphocytes, and endothelial cell,s and obtained 210,518 PPIs involving 8,635 proteins. The team created a public database with a web interface to download, search, and visualize these PPIs (https://genemo.ucsd.edu /proper).
|
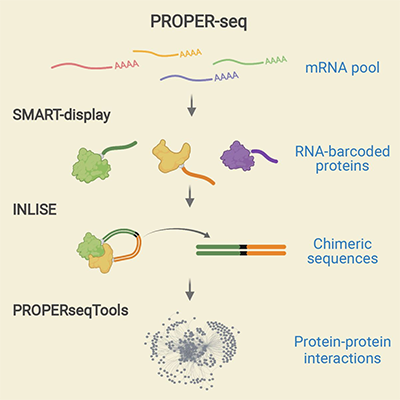 |
| A graphical abstract of the three tools that together are known as PROPER-seq.
|
|
The team validated the PROPER-seq-identified PPIs (called PROPER v1.0) with previously characterized PPIs documented in PPI databases. The team found more than 1,300 and 2,400 PPIs in PROPER v1.0 are supported by previous co-immunoprecipitation experiments and affinity purification-mass spectrometry experiments, respectively.
|
|
The team experimentally validated four PROPER-seq identified PPIs that have not been reported in the literature. These four PPIs involve PARP1, a critical protein for DNA repair and a drug target of several human cancers, and four other proteins involved in the trafficking of molecules and transcription regulation. These validations suggest mechanistic links between PARP1 and import/export of molecules to/from the nucleus as well as gene transcription.
|
|
Their results show that PROPER v1.0 overlaps with more than 17,000 computationally predicted PPIs without prior experimental validation. The experimental support offered by PROPER-seq to these previously uncharacterized PPIs suggests the solid predictive ability of protein structure-based computational models.
|
|
The team found PROPER v1.0 overlaps with one hundred synthetic lethal (SL) gene pairs. An SL gene pair can cause cell death when both genes of this gene pair are lost. This finding suggests a connection between physical interactions (PPIs) and human genetic interactions.
|
|
Looking forward, the team hopes PROPER-seq can assist researchers in screening many protein pairs and identify PPIs of interest. In addition, the PROPER-seq identified PPIs from different labs can expand the PPI networks' reference maps and illuminate cell-type-specific PPIs.
|

