Western Blot / Immunoblot: Definition, Workflow, Uses, and Limitations
What Is a Western Blot?
A Western blot, also called an immunoblot, is a laboratory technique used to detect a specific protein-reactive band within a complex mixture by combining size-based electrophoretic separation with antibody-based detection. After proteins are separated by gel electrophoresis, they are transferred – or “blotted” – onto a thin membrane, where an antibody binds the target-associated band and produces a visible or digital signal.
In simple terms: a Western blot separates proteins by size, transfers them to a membrane, and uses antibodies to reveal whether a target protein appears at the expected apparent molecular weight. It is best used as evidence for protein presence, size, and relative abundance, not as a standalone proof of molecular identity or an inherently precise concentration assay.
Synonyms: immunoblot, protein blot, Western blotting.
Not to be confused with: Southern blot, which detects DNA, or Northern blot, which detects RNA.
Key points: A Western blot can show whether a target protein is detectable, whether the detected band appears near the expected molecular weight, and – when performed within a validated linear range – how its abundance compares across samples on the same blot. The technique is widely used to confirm protein expression, validate the activity of recombinant proteins, monitor post-translational modifications such as phosphorylation, and verify that an antibody recognizes its intended target across biotechnology research and industrial applications.
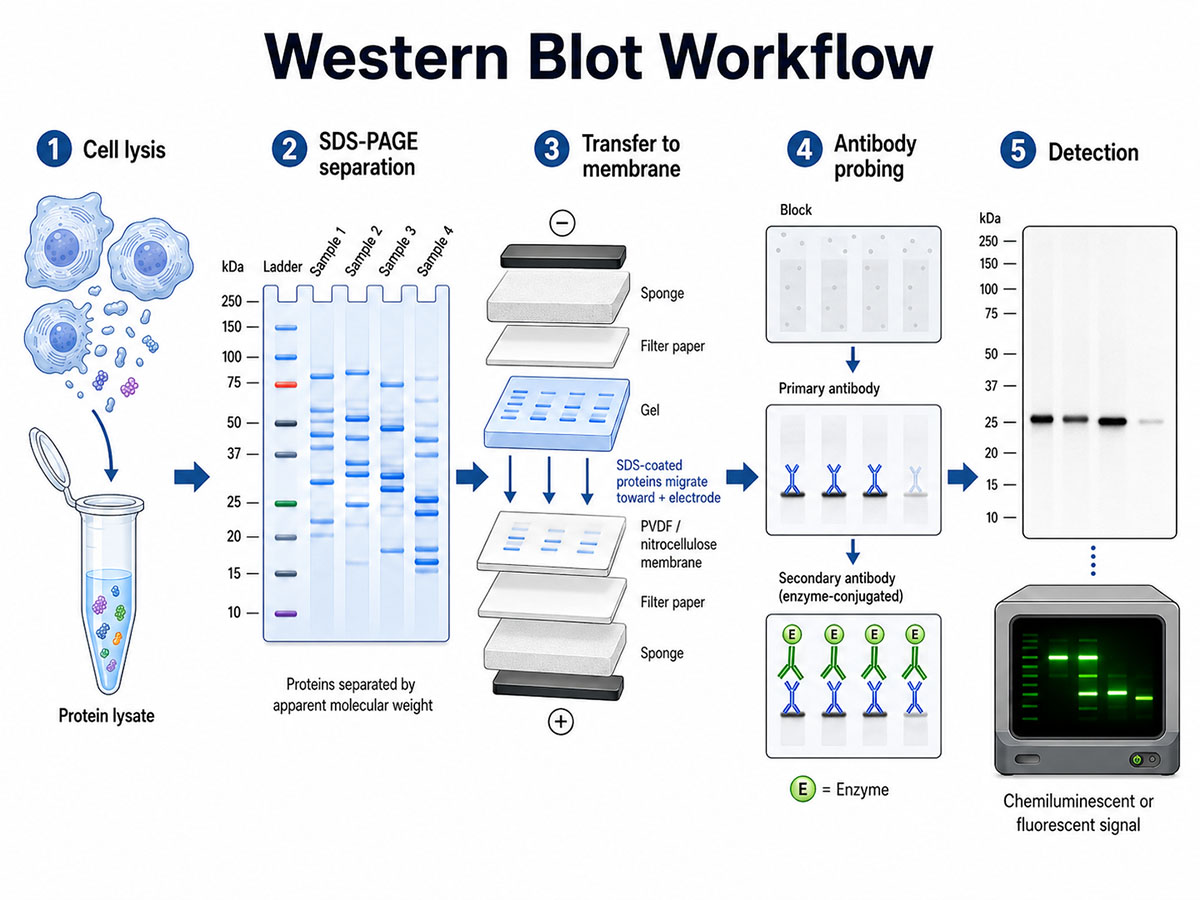
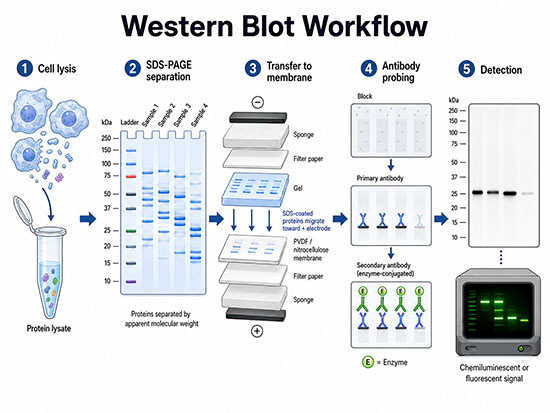
The Western blot workflow in five stages: (1) cell lysis and protein lysate preparation; (2) SDS-PAGE separation by apparent molecular weight; (3) electrophoretic transfer of SDS-coated proteins from the gel to a PVDF or nitrocellulose membrane toward the positive electrode; (4) blocking followed by primary and enzyme-conjugated secondary antibody incubation; and (5) chemiluminescent or fluorescent detection of the target band. (Image: Nanowerk) (click on image to enlarge)
What a Western Blot Can and Cannot Tell You
A well-designed Western blot can provide three kinds of evidence: whether an antibody-reactive band is detectable, whether that band migrates near the target protein's expected apparent molecular weight, and whether the signal changes between samples run and detected under comparable conditions. These strengths make the method especially useful for confirming protein expression, checking cleavage products or isoforms, and validating whether an antibody behaves as expected in a given sample type.
A Western blot cannot, by itself, prove a protein's molecular identity, measure an absolute concentration without calibration, or exclude all off-target antibody binding. A convincing result usually depends on controls: a positive sample, a negative or knockout/knockdown sample where feasible, a molecular-weight marker, a loading control or total-protein stain, and exposure conditions that keep the signal inside the linear detection range.
History of the Western Blot
The technique emerged in 1979 from two independent groups. Jaime Renart, Jakob Reiser, and George Stark, working at Stanford University, published in PNAS in July a method that transferred proteins from polyacrylamide gels onto chemically activated diazobenzyloxymethyl paper for antibody detection. Two months later, Harry Towbin, Theophil Staehelin, and Julian Gordon at the Friedrich Miescher Institute in Basel published the variant that used unmodified nitrocellulose, was simpler to perform, and used a labeled secondary antibody for detection. The Towbin nitrocellulose protocol is the one that became dominant. W. Neal Burnette gave the technique its modern name in a 1981 paper in Analytical Biochemistry, by analogy to the Southern blot for DNA (Edwin Southern, 1975) and the Northern blot for RNA (1977). By the early 2010s, immunoblotting was standard in cell biology and molecular biology laboratories.
How Does a Western Blot Work?
A Western blot proceeds through five sequential steps: sample preparation, electrophoretic separation, transfer to a membrane, antibody probing, and detection. Small variations in any of these can explain why Western blots from different laboratories sometimes disagree on the same target. Exact times, voltages, antibody dilutions, blocking conditions, and sample preparation choices vary by protein, antibody, membrane, and instrument.
Sample preparation begins with cell or tissue lysis in a buffer containing detergents, protease inhibitors, and, for phosphorylation studies, phosphatase inhibitors. Total protein concentration is usually measured by a colorimetric assay such as the BCA or Bradford method so that comparable amounts of protein can be loaded into each gel well. The lysate is then mixed with sample buffer containing sodium dodecyl sulfate (SDS), a reducing agent such as β-mercaptoethanol or dithiothreitol, glycerol, and a tracking dye, then usually heated to denature proteins and coat them with negative charge. Heating conditions vary: some membrane proteins, aggregates, and multi-pass proteins require gentler treatment to avoid aggregation or loss of signal.
In the electrophoresis step, the prepared samples are loaded into the wells of a polyacrylamide gel and an electric field drives them through the gel matrix. Because SDS gives most proteins a broadly similar mass-to-charge ratio, smaller proteins usually migrate faster and larger ones migrate more slowly, separating the mixture into bands by apparent molecular weight. Gel acrylamide concentration is chosen for the target’s size, with lower-percentage gels favoring large proteins and higher-percentage or gradient gels improving resolution of smaller proteins. This step, known as SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis), gives Western blotting its size-based check on antibody detection.
The third step transfers the separated proteins from the fragile gel onto a more durable membrane, originally nitrocellulose and now most often polyvinylidene difluoride (PVDF) with a pore size of 0.2 or 0.45 µm. Transfer can be performed by wet blotting, in which the gel-membrane sandwich is fully submerged in transfer buffer and run under conditions chosen for the target size and membrane, or by semi-dry blotting, which is faster but can be less efficient for large proteins. PVDF is often chosen over nitrocellulose when higher protein-binding capacity, chemical resistance, or stripping and reprobing are important.
Once the proteins are immobilized on the membrane, free binding sites are blocked with a non-specific protein solution, typically 5% non-fat dry milk or bovine serum albumin in Tris-buffered saline with 0.1% Tween-20. The membrane is then incubated with a primary antibody – diluted under antibody-specific conditions and often incubated overnight at 4 °C – that binds the target protein. After washing, the membrane is incubated with a secondary antibody conjugated to a reporter enzyme such as horseradish peroxidase or alkaline phosphatase. The two-antibody arrangement amplifies signal, since multiple labeled secondaries can bind each primary.
Detection is performed by adding a substrate that produces a chemiluminescent or fluorescent signal where the antibodies have bound. The signal is captured on X-ray film or, increasingly, on a digital imager, producing the dark bands that are the familiar visual output of a Western blot. The position of each band relative to a molecular-weight ladder estimates the apparent molecular weight of the detected signal and helps determine whether it appears near the expected size. Band intensity can be analyzed by densitometry to compare protein levels between samples when the signal is captured within the assay's linear range.
How to Interpret Western Blot Bands
A Western blot is interpreted by asking whether the observed band pattern matches the biological expectation. A single band near the predicted apparent molecular weight is usually easier to interpret than multiple bands, but additional bands do not automatically mean the experiment failed. They may reflect isoforms, cleavage products, glycosylation, phosphorylation-dependent mobility shifts, dimers, degradation, or non-specific antibody binding. Conversely, a band at the expected size is not definitive proof of identity unless the antibody and sample controls support that interpretation.
For relative quantification, the most important question is whether the detected signal is unsaturated and proportional to the amount of protein loaded. Overexposed chemiluminescent bands can look persuasive while being unusable for densitometry. Quantitative claims are strongest when they are based on biological replicates, linear-range exposure, consistent normalization, and analysis of samples run on the same membrane or linked by a shared standard.
Variants of the Western Blot
Several related techniques modify or extend the standard Western blot. The dot blot skips electrophoresis entirely and applies the protein sample directly to the membrane as a spot, which is faster but provides no size information and cannot distinguish the target from cross-reacting bands. The far-Western blot replaces the antibody with another protein – for example a candidate binding partner – to detect protein-protein interactions on the membrane. In a native Western blot, proteins are separated without SDS denaturation, helping preserve complexes or conformational epitopes. In a two-dimensional Western blot, proteins are first separated by isoelectric point and then by molecular weight, improving resolution of isoforms or post-translationally modified variants.
Since the late 2000s, capillary Western platforms have emerged as an alternative to traditional gel-and-membrane formats. Commercial systems such as ProteinSimple’s Wes, Jess, and Abby instruments separate proteins in capillaries, immobilize them, and automate antibody incubation, washing, and detection, requiring small sample volumes and producing more standardized, readily quantifiable results in roughly three hours. The trade-off is high instrument and reagent cost, and quantification still depends on antibody specificity, calibration, dynamic range, and assay validation.
Multiplex Western blots use spectrally distinct fluorescent secondary antibodies to detect two or more targets on the same membrane simultaneously. This is useful for tracking modified and unmodified forms of the same protein side by side – for example phosphorylated and total ERK kinase – or for measuring a target and its loading control in a single image.
Western Blot Versus ELISA and Mass Spectrometry
When a researcher needs to know whether a particular protein is present in a sample, several complementary techniques may apply. The Western blot, ELISA, and mass spectrometry overlap in capability but differ markedly in throughput, sensitivity, and specificity, and each occupies a distinct niche in the laboratory.
| Feature | Western blot | ELISA | Mass spectrometry |
|---|---|---|---|
| Separation principle | Size separation in gel, then antibody binding | Antibody capture in microplate well | Mass-to-charge separation of peptides |
| Output | Bands at known molecular weight | Quantitative concentration | Peptide identity and abundance |
| Throughput | Low (1–2 days, 10–15 samples per blot) | High (hours, 96- or 384-well plates) | Moderate to high, depending on platform |
| Sensitivity | Variable; often low-picogram to nanogram depending on antibody and detection chemistry | Very high (picogram to femtogram) | Very high but matrix-dependent |
| Quantification | Semi-quantitative | Quantitative within a validated standard-curve range | Quantitative with internal standards |
| Antibody required | Yes | Yes | No (but can be combined with antibody enrichment) |
The non-obvious advantage of the Western blot is that the size separation visually exposes antibody mistakes. If a primary antibody binds an off-target protein of the wrong molecular weight, the band appears in the wrong place – an error an ELISA, which reads only signal intensity in a well, would silently include in its reported concentration. ELISA delivers higher throughput and quantitative concentration measurements but cannot distinguish the target from a cross-reactive impurity. Mass spectrometry can identify proteins at the peptide level and does not require an antibody, but protein inference, peptide detectability, sample preparation, instrument access, and method-development costs make it a different kind of assay rather than a simple replacement for immunoblotting. The three techniques are often used together: Western blot to identify, ELISA to quantify, and mass spectrometry for unbiased proteomics or to confirm antibody specificity.
Applications in Research and Clinical Diagnostics
Western blotting underpins routine work across cell biology, biochemistry, and translational medicine. In research, it is the standard method for confirming a gene knockout or knockdown, measuring how protein levels change in response to a treatment, verifying that a transfected construct produces a protein of the expected size, and tracking post-translational modifications such as phosphorylation, ubiquitination, or proteolytic cleavage that shift a band’s position or intensity. The technique is also a workhorse of biomarker validation, where a candidate identified by mass spectrometry or transcriptomics is confirmed by antibody-based detection in patient samples. Where suitable antibodies exist, Western blotting is often a relatively inexpensive way to ask whether a target-associated band appears under defined experimental conditions.
In biopharmaceutical manufacturing, Western blotting can support characterization of therapeutic recombinant proteins and monoclonal antibodies. It can help confirm the size and identity of an active ingredient, detect host cell protein contaminants from CHO or E. coli production lines, and check that a molecule has not been clipped or aggregated during purification – orthogonal characterization that may support regulatory submissions alongside assays such as ELISA, capillary electrophoresis, and mass spectrometry in biopharmaceutical development.
The technique’s role in clinical diagnostics has narrowed substantially over the past decade. From the late 1980s through 2014, Western blot was widely used as the standard supplemental confirmatory test for HIV antibodies in the United States, and a positive Western blot became cultural shorthand for a confirmed HIV diagnosis. In 2014 the U.S. Centers for Disease Control and Prevention replaced this algorithm with one in which positive HIV screening tests are confirmed by an HIV-1/HIV-2 antibody differentiation immunoassay rather than by Western blot, because the newer assay detects acute infections earlier and removes some of the indeterminate results that complicated Western blot interpretation. Western blot/immunoblot remains part of the traditional standard two-tier serologic algorithm for Lyme disease, where positive or equivocal first-tier enzyme immunoassays are followed by IgG and IgM immunoblots against Borrelia burgdorferi antigens, although modified two-tier approaches using sequential enzyme immunoassays are now also used.
Limitations and the Reproducibility Challenge
Despite its prevalence, Western blot has acknowledged weaknesses, and the conversation about how to fix them has grown louder over the past decade. The technique is fundamentally semi-quantitative: signal intensity has a non-linear relationship with protein amount, the linear dynamic range of chemiluminescent detection typically spans only one to two orders of magnitude (compared with four or more for fluorescent detection on a CCD imager), and band intensity depends on transfer efficiency, antibody concentration, exposure time, and detector linearity in ways that vary between blots. Recent methodology papers argue that obtaining genuinely quantitative data requires standard curves derived from serial dilutions, multiple replicates, and rigorous attention to detector linearity that most published Western blots lack.
Antibody quality is one of the largest sources of unreliable Western blot results. A substantial fraction of commercial primary antibodies have not been adequately validated, and unvalidated antibodies routinely produce bands at the wrong molecular weight or fail to detect the target at all. Editorial guidelines from major journals increasingly ask authors to provide antibody details, validation information, full blot images, and, where appropriate, knockdown or knockout controls when reporting Western blot results.
Normalization remains a methodological flashpoint. The traditional approach divides the target band’s intensity by that of a “housekeeping” protein such as GAPDH, β-actin, or α-tubulin, on the assumption that these are expressed at equal levels across samples. Recent comparative studies have shown that housekeeping protein abundance varies meaningfully across cell types, treatments, and disease states. Total protein staining methods, in which the entire protein content of each lane is stained with a fluorescent dye and used as the loading reference, generally produce lower coefficients of variation across replicates, and many methodologists and some journal guidelines now encourage total protein normalization, especially for quantitative claims.
Common pitfalls include overloaded lanes, incomplete transfer of large proteins, saturated chemiluminescent exposures, non-specific antibody bands, unstable housekeeping-protein controls, and comparing bands from different membranes without a shared standard. Good practice includes using validated antibodies, molecular-weight markers, positive and negative controls, biological replicates, full-blot images, and normalization within the linear range of detection. Essential controls include a positive control, a negative or knockout/knockdown control, a molecular-weight ladder, a loading control or total-protein stain, and, for modified proteins, a control showing that the modification-specific antibody is selective.
Throughput and cost are also constraints. A typical Western blot takes one to two days from lysis to imaging, a single conventional mini-gel membrane usually accommodates roughly 10 to 15 samples, and consumables for membranes, antibodies, and detection reagents add up over a project. Large-scale protein expression studies therefore tend to use proteomic mass spectrometry, with Western blots reserved for validating the targets that emerge.
Future Directions
Three trends are reshaping how Western blots are run. Automated capillary platforms are making immunoblotting more standardized and easier to quantify, at the cost of higher reagent prices and proprietary instruments. As biopharmaceutical manufacturers and core facilities adopt these systems, expectations for reproducible Western blot data are rising.
Better validation pipelines are reducing the antibody-quality problem at its source. Initiatives such as the YCharOS consortium and the Antibody Registry are systematically validating commercial antibodies against tagged endogenous proteins or knockout cell lines, and journal pressure to cite RRID identifiers makes it easier to track which reagents have been independently verified. Recombinant antibodies and nanobodies, which are sequence-defined and reproducible batch to batch through protein engineering, are gradually replacing hybridoma-derived monoclonals in critical applications.
Single-cell Western blotting, still more specialized than routine immunoblotting, extends the technique’s specificity advantages to individual cells rather than bulk lysates. Combined with multiplex detection and machine-learning-assisted band quantification, these advances may keep Western blotting central to protein analysis even as proteomic mass spectrometry continues to expand.
Frequently Asked Questions
Why is it called a Western blot? The name is a deliberate play on the Southern blot, a DNA detection technique invented by British biologist Edwin Southern in 1975. By analogy, the RNA version became the Northern blot, and W. Neal Burnette gave the protein version the name Western blot in his 1981 paper. The naming joke does not refer to any geographic feature – all three blots can be performed anywhere.
What is the difference between a Western blot and an ELISA? Both techniques use antibodies to detect specific proteins, but they answer slightly different questions. An ELISA captures the target in a microplate well and gives a quantitative concentration but no size information, so an antibody binding a cross-reactive impurity would not be flagged by molecular-weight information. A Western blot first separates proteins by size on a gel before antibody detection, allowing visual confirmation that the band appears at the correct molecular weight. The two assays are often performed on the same samples.
Is the Western blot still used to diagnose HIV? Not in the standard U.S. testing algorithm. Since 2014 the U.S. Centers for Disease Control and Prevention has recommended an HIV-1/HIV-2 antibody differentiation immunoassay, rather than Western blot, to confirm a positive screening result, because the newer algorithm detects acute infections earlier and produces fewer indeterminate results. Western blot/immunoblot remains part of the traditional standard two-tier algorithm for Lyme disease, though modified two-tier approaches using sequential enzyme immunoassays are also used.
Can a Western blot membrane be stripped and reprobed? Yes, and this is a common practical use of the technique. After detection, the bound primary and secondary antibodies can be removed by incubating the membrane in a mild stripping buffer – typically a low-pH glycine solution or a commercial reagent – allowing a different antibody to be applied to the same membrane. PVDF often tolerates stripping and reprobing better than nitrocellulose, which is one reason many laboratories choose it for experiments that require repeated probing. Each strip cycle reduces the remaining protein on the membrane and increases background, so most laboratories limit a single membrane to two or three rounds of probing.
How do I know my antibody is detecting the right protein? The most reliable way is to include a knockout or knockdown control – a sample in which the target gene has been disrupted – and confirm that the band of interest disappears. Other approaches include comparing the band’s molecular weight to the predicted size of the target, checking that overexpression of a tagged version produces a stronger band, and consulting independent validation data from initiatives such as YCharOS or the Human Protein Atlas. Many commercial antibodies have not been adequately validated, so this confirmation step is essential.
Further Reading
Proceedings of the National Academy of Sciences, Electrophoretic Transfer of Proteins from Polyacrylamide Gels to Nitrocellulose Sheets: Procedure and Some Applications
Proceedings of the National Academy of Sciences, Transfer of Proteins from Gels to Diazobenzyloxymethyl-Paper and Detection with Antisera: A Method for Studying Antibody Specificity and Antigen Structure
Analytical Biochemistry, “Western Blotting”: Electrophoretic Transfer of Proteins from Sodium Dodecyl Sulfate–Polyacrylamide Gels to Unmodified Nitrocellulose and Radiographic Detection with Antibody and Radioiodinated Protein A
Scientific Reports, A Critical Path to Producing High Quality, Reproducible Data from Quantitative Western Blot Experiments
Methods and Protocols, Comparison of Automated and Traditional Western Blotting Methods
