| Jun 27, 2023 |
Researchers develop new base editing tools using AI-predicted protein structure clustering
|
|
(Nanowerk News) AO Caixia's group from the Institute of Genetics and Developmental Biology of the Chinese Academy of Sciences has pioneered the use of artificial intelligence (AI)-assisted methods to discover novel deaminase proteins with unique functions through structural prediction and classification.
|
|
This approach has opened up a range of applications for the discovery and creation of desired plant genetic traits.
|
|
The results were published in Cell ("Discovery of deaminase functions by structure-based protein clustering").
|
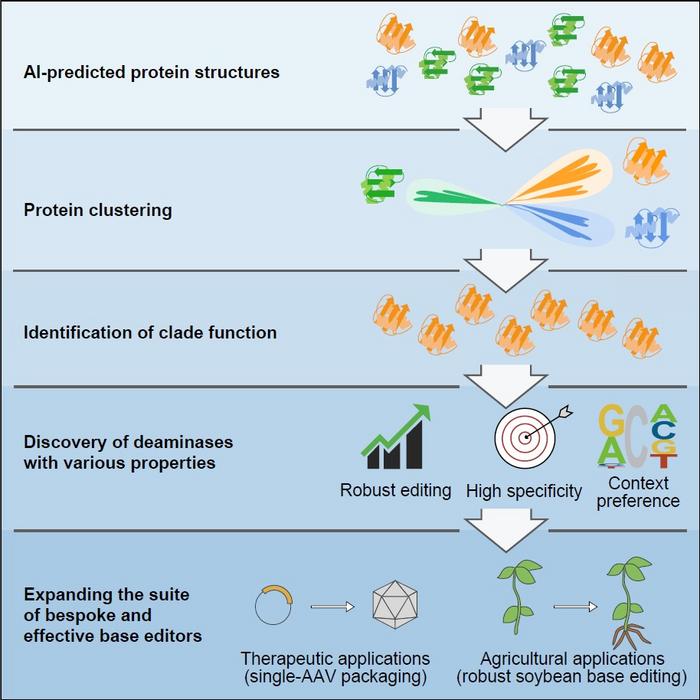 |
| AI-assisted structural predictions and alignments establishes a new protein classification and functional mining method, further enabling the discovery of a suite of single and double-strand cytidine deaminases which show great potential as bespoke base editors for therapeutic or agricultural breeding applications. (Image: IGDB)
|
|
The discovery of new proteins and the exploitation of diverse engineered enzymes have contributed to the rapid advancement of biotechnology. Currently, efforts to mine novel proteins generally rely on amino acid sequences, which cannot provide a robust link between protein structural information and function.
|
|
Base editing is a new precision genome editing technology that has the potential to revolutionize molecular crop breeding by introducing desired traits into elite germplasm. The discovery of several deaminases has expanded the capability of cytosine base editing. Although traditional sequence-based efforts have identified many proteins for use as base editors, limitations in editing specific DNA sequences or species still remain.
|
|
Canonical efforts based solely on protein engineering and directed evolution have helped to diversify base editing properties, but challenges persist. By predicting the structures of proteins within the deaminase protein family using AlphaFold2, the researchers clustered and analyzed deaminases based on structural similarities. They identified five new deaminase clusters with cytidine deamination activity in the context of DNA base editors.
|
|
Using this approach, they further reclassified a group of cytidine deaminases, called SCP1.201 and previously thought to act on dsDNA, to perform deamination primarily on ssDNA. Through subsequent protein profiling and engineering efforts, they developed a suite of new DNA base editors with remarkable features. These deaminases exhibit properties such as higher efficiency, lower generation of off-target editing events, editing at different preferred sequence motifs, and much smaller size.
|
|
The researchers emphasized that the development of a suite of base editors would enable future tailor-made applications for various therapeutic or agricultural breeding efforts. They developed the smallest single-strand specific cytidine deaminase, enabling the first efficient cytosine base editor to be packaged in a single adeno-associated virus.
|
|
They also discovered a highly effective deaminase from this clade specifically for soybean plants, a globally significant agricultural crop that previously exhibited poor editing by cytosine base editors.
|
|
In general, the recent advent of protein structure prediction using growing genomic databases will greatly accelerate the development of new bioengineering tools.
|
|
This study highlights an approach that uses just the cytidine deaminase superfamily to develop a suite of new technologies and uncover new protein functions. These newly discovered deaminases, based on AI-assisted structural predictions, greatly expand the utility of base editors for therapeutic and agricultural applications.
|
|
In addition, this study will be of broad interest to the larger research community in phylogenetics, metagenomics, protein engineering and evolution, genome editing, and plant breeding.
|

