| Posted: Sep 16, 2014 |
The extended stability range of phosphorus allotropes made of nanotubes
|
|
(Nanowerk News) What holds white, black, and red phosphorus together—and prevents it from falling apart, for example into much-sought-after atomically thin networks and nanowires? This is what German scientists now found out using numerical modeling. As they explain in the journal Angewandte Chemie ("The Extended Stability Range of Phosphorus Allotropes"), Van der Waals forces, weak interactions between covalently bonded phosphorus units, play the key role.
|
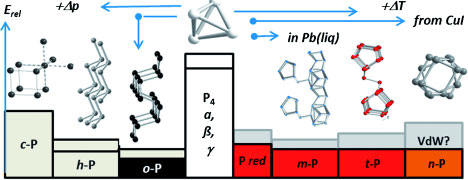 |
| Computation of the stabilities and crystal structures of known and new phosphorus allotropes made of nanotubes. (©Wiley-VCH)
|
|
Phosphorus occurs in several different forms—more than those described in classic textbooks. They each consist of different molecular building blocks aggregated into a solid. Reactive, white phosphorus is used in matchsticks, among other things, and exists in three different crystal structures. These all consist of individual tetrahedra made of phosphorus atoms, but the tetrahedra are arranged differently within the different crystal structures.
|
|
Under pressure—or by means of a new method developed by Tom Nilges (TU Munich) and Peer Schmidt (BTU Cottbus-Senftenberg)—white phosphorus can be converted to inert black phosphorus, the most stable form at room temperature. This form and another high-pressure allotrope both consist of corrugated layers of phosphorus atoms. The isolation of individual layers to make “phosphorene” (analogous to graphene) is the object of current research. When white phosphorus is heated, it is converted to various forms of red phosphorus.
|
|
Until recently, only the structure of one of these, violet Hittorf’s phosphorus, was known. It consists of pentagonal nanotubes of covalently bonded phosphorus atoms. In recent years, Arno Pfitzner (University of Regensburg), Michael Ruck (TU Dresden), and other researchers have produced other allotropes containing single and paired phosphorus nanotubes that occur as fibrous red crystals or red-brown variants. The structures of the nanotube modifications and the stabilities of the new forms relative to the known ones remained unclear.
|
|
Richard Weihrich, Arno Pfitzner, and their co-workers at the University of Regensburg, RWTH Aachen, BTU Cottbus-Senftenberg, and TU Munich have now successfully used density functional theory (DFT) to establish the order of stability for the entire series of crystalline phosphorus allotropes and to determine the structures of the new single-rod modifications.
|
|
Although conventional DFT calculations were too imprecise, the researchers were able to obtain excellent agreement with experimental results and precise predictions by using a special correction term. This term takes into consideration the Van der Waals forces, weak interactions between the molecules, layers, and tubes of the different forms. These interactions play a significant role for phosphorus.
|
|
“Despite significant differences, all phosphorus allotropes consist of covalent substructures that are held together by Van der Waals interactions,” explains Weihrich. “We have now been able to comprehensively compute the stabilities of even those allotropes that are energetically very similar. We have also been able to correctly predict structures on the basis of the van der Waals interactions. It was thus also possible to integrate the recently discovered tubular modifications and to predict the structure of the previously unknown crystalline structures of phosphorus nanotubes.”
|
|
These weak interactions are highly relevant to new research into single- and multiple-layer “phosphorenes” as well as the possible separation of individual phosphorus nanotubes.
|

