| Sep 17, 2020 |
Self-imaging of a molecule by its own electrons: mapping the atomic motion during a molecular vibration(Nanowerk News) One of the long-standing goals of research on the light-induced dynamics of molecules is to observe time-dependent changes in the structure of molecules, which result from the absorption of light, as directly and unambiguously as possible. To this end, researchers have developed and applied a plethora of approaches. |
| Of particular promise among these approaches are several methods developed in the last years that rely on diffraction (of light or electrons) as means of encoding the internuclear spacings between the atoms that together form the molecule. |
| In a recent paper in Physical Review Letters ("Evolution of a Molecular Shape Resonance Along a Stretching Chemical Bond"), researchers at the MBI led by Dr. Arnaud Rouzée have shown that high-resolution movies of molecular dynamics can be recorded using electrons ejected from the molecule by an intense laser field. |
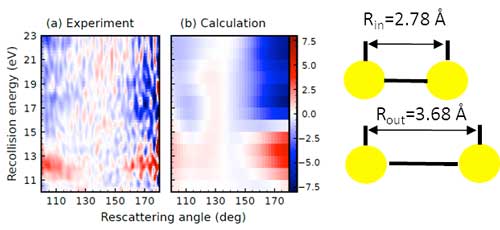 |
| Fig. 1: Difference between the electron scattering cross-section measured (a) and calculated (b) at R = 3.68 Å (corresponding to the outer turning point of the vibrational motion) and R = 2.78 Å (corresponding to the inner turning point of the vibrational motion) for the case of an I2 vibrational wave packet created by photo-excitation of the B-state using a visible laser with a wavelength of 555 nm. The difference in the scattering cross-sections is shown as a function of the kinetic energy of the returning electron and the angle into which the electron is scattered. Particularly near a rescattering angle of 180 degrees (i.e. for back-scattered electrons) a major difference is seen between the scattering cross-section at the geometry corresponding to the inner and outer turning point of the vibration. In other words, the time-dependent changes in the internuclear distance are recognizable in time-dependent changes of the measured electron scattering cross-sections. (Image courtesy of the researchers) |
| Following strong field ionization, the electrons that are set free are generally accelerated away from the molecule under the influence of the laser electric field. |
| However, due to the oscillating nature of this field, a fraction of the electrons are driven back to their parent molecular ion. |
| This sets the stage for a so-called re-collision process, in which the electron can be reabsorbed in the molecule (and where the absorbed energy is released in the form of high energy photons) or scatters off the molecular ion. Depending on the kinetic energy of the electron, it can be transiently trapped inside a centrifugal potential barrier. |
| This is a well-known process in electron scattering and in single photon ionization experiments, and is referred to as a shape resonance. The smoking gun for the occurrence of a shape resonance is a large increase of the scattering cross-section. As its name implies, the kinetic energy for which the shape resonance occurs is highly sensitive to the shape of the molecular potential, and consequently to the molecular structure. |
| Therefore, shape resonances can be used to make a movie of a molecule that is undergoing ultrafast nuclear rearrangement. |
| To demonstrate this effect, the team at MBI recorded a movie of the ultrafast vibrational dynamics of photo-excited I2 molecules. |
| A first laser pulse, with a wavelength in the visible part of the wavelength spectrum, was used to prepare a vibrational wavepacket in the electronic B-state of the molecule. This laser pulse was followed by a second, very intense, time-delayed laser pulse, with a wavelength in the infrared part of the wavelength spectrum. |
| Electron momentum distributions following strong field ionization by the second laser pulse were recorded at various time delays between the two pulses, corresponding to different bond distances between the two iodine atoms. A strong variation of the laser-driven electron rescattering cross-section was observed with delay, which could unambiguously be assigned to a change of the shape resonance energy position (see Fig. 1) induced by the vibrational wavepacket motion. |
| As such, this work introduces new opportunities for investigating photo-induced molecular dynamics with both high temporal and spatial resolution. |
| Source: Forschungsverbund Berlin |
|
Subscribe to a free copy of one of our daily Nanowerk Newsletter Email Digests with a compilation of all of the day's news. |

