Quantum Chemistry: Definition, Methods, and Nanoscience Applications
What is Quantum Chemistry?
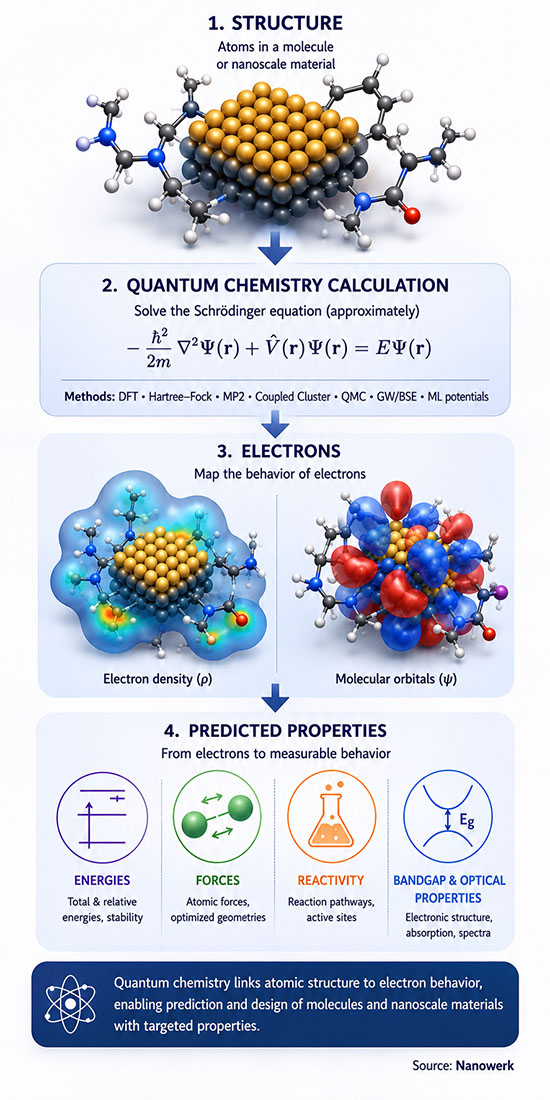
Quantum chemistry applies quantum mechanics to atoms, molecules, and materials to predict electronic structure, energies, geometries, spectra, and reactivity. Most practical calculations use approximations to the Schrödinger equation, ranging from first-principles methods to parameterized models.
In simple terms, quantum chemistry uses physics-based calculations to predict how electrons arrange themselves. That electron arrangement determines how atoms bond, how molecules react, how materials absorb light, and why nanoscale structures often behave differently from bulk matter.
Key takeaways:
- Quantum chemistry predicts molecular and materials properties from electronic structure.
- Its methods form a hierarchy from fast approximate models to high-accuracy wavefunction calculations.
- The best method depends on the system size, the property of interest, and the required accuracy.
- It is central to nanoscience because nanoscale optical, electronic, magnetic, and catalytic behavior is governed by quantum electronic states.
In a quantum-chemical calculation the basic objects are not atoms drawn as spheres but electrons around nuclei, represented either by a many-electron wavefunction or, in density functional theory, by the electron density. These quantities are used to compute bond lengths and angles, dissociation energies, vibrational spectra, optical absorption, magnetic response, dipole moments, reaction barriers, and the relative stability of competing structures. In practice, many properties also require additional approximations, such as response theory for spectra, transition-state searches for reaction barriers, statistical sampling for temperature effects, or solvent and surface models for realistic environments.
The field began in 1927, the year after Schrödinger published his equation, when Walter Heitler and Fritz London showed that a chemical bond emerges from the quantum mechanics of two electrons in the hydrogen molecule. Later work produced Hartree-Fock theory, configuration-interaction expansions, basis sets for molecules, band theory for solids, and pseudopotentials for heavy atoms and periodic materials. A major turning point came in 1964–1965 with the Hohenberg-Kohn theorems and the Kohn-Sham equations, which made density functional theory the workhorse of modern electronic-structure calculations. Today quantum chemistry is overwhelmingly computational and is used to interpret experiments, design catalysts, and screen candidate materials at the nanoscale before they are synthesized.
The Quantum-Mechanical Foundation
The starting point is the time-independent Schrödinger equation for a molecule or solid, which contains kinetic energy operators for all electrons and nuclei together with electron-nuclear, electron-electron, and nuclear-nuclear Coulomb interactions. Two simplifications make the problem tractable. The Born-Oppenheimer approximation separates electronic and nuclear motion by exploiting the fact that nuclei are at least 1836 times heavier than electrons, so the electrons relax effectively instantaneously to any nuclear configuration. The non-relativistic limit is adopted for light elements, while heavy elements are treated with relativistic corrections or pseudopotentials that absorb core-electron physics.
After these simplifications the central object is the many-electron wavefunction, which depends on the spatial and spin coordinates of every electron. Because electrons are identical fermions, the wavefunction must change sign under exchange of any two of them, a constraint that gives rise to the Pauli exclusion principle and to the antisymmetric Slater-determinant ansatz that underlies most wavefunction methods. The variational principle guarantees that any trial wavefunction yields an energy bounded from below by the true ground-state energy, which sets the optimization target for almost every quantum-chemical method. The wave nature of electrons, captured by the de Broglie wavelength, is what makes quantum effects dominant below a few nanometers. Solving these equations exactly is impossible for any system of practical interest because the exact wavefunction lives in a Hilbert space whose dimension grows exponentially with electron number, so the history of quantum chemistry is the history of approximations that capture as much of the true wavefunction or density as possible at polynomial cost.
Approximation Methods in Quantum Chemistry
Quantum-chemical methods cluster into a few families that differ in how they treat the central problem of electron correlation, the cooperative motion of electrons that arises from their mutual repulsion. The right method depends on the size of the system, the property of interest, and the accuracy required.
Hartree-Fock and post-Hartree-Fock methods
Hartree-Fock theory replaces the full many-electron wavefunction with a single Slater determinant of one-electron orbitals and treats electron repulsion only on average through a self-consistent mean field. It captures roughly 99 percent of the total electronic energy but typically misses about 1 percent that, in chemical terms, sets the scale of bond energies and reaction barriers. This residual is called the correlation energy. Post-Hartree-Fock methods recover correlation by expanding the wavefunction in additional Slater determinants. Møller-Plesset perturbation theory, configuration interaction, multireference approaches, and especially coupled-cluster theory form a hierarchy of increasing accuracy and increasing cost. Coupled cluster with single, double, and perturbative triple excitations — CCSD(T) — is widely regarded as a "gold standard" for single-reference systems, reaching chemical accuracy of roughly 1 kcal/mol on equilibrium energies, but its cost scales as the seventh power of system size and limits routine application to a few dozen atoms.
Density functional theory
Density functional theory shifts the fundamental variable from the many-electron wavefunction to the electron density, a function of just three spatial coordinates. The Hohenberg-Kohn theorems prove that the ground-state density determines all properties of the system; the Kohn-Sham equations recast the problem as a system of non-interacting electrons moving in an effective potential that includes a so-called exchange-correlation functional. The exact functional is unknown, so practical DFT calculations rely on approximations of escalating sophistication, often visualized as a "Jacob's ladder" running from the local density approximation through generalized-gradient approximations, meta-GGAs, hybrid functionals that mix in exact exchange, and double-hybrid functionals that add perturbative correlation. Modern DFT often scales roughly as the third power of system size and can treat hundreds to a few thousand atoms when the functional, basis, and implementation are suitable; linear-scaling implementations can push this further. It is the dominant method in materials science, surface science, and much of nanoscience, but its accuracy depends strongly on the exchange-correlation functional and on the type of bonding or excitation being studied.
Quantum Monte Carlo and many-body Green's-function methods
Quantum Monte Carlo methods sample the many-electron wavefunction stochastically and can deliver near-exact ground-state energies for systems of up to a few hundred electrons, at high computational cost. Diffusion Monte Carlo and variational Monte Carlo are particularly useful for benchmarking DFT and for strongly correlated systems where coupled cluster is unreliable. Many-body Green's-function approaches, including the GW approximation for charged excitations and the Bethe-Salpeter equation for neutral excitations, give accurate quasiparticle bandgaps and optical absorption spectra of solids and nanostructures and are the methods of choice when precise bandgap values matter.
Semiempirical and tight-binding methods
Semiempirical methods such as AM1, PM7, and the modern GFN-xTB family retain a quantum-mechanical framework but replace many of the difficult integrals with parameters fitted to experiment or higher-level calculations. Tight-binding and density-functional tight-binding extend this idea to large solids and nanostructures. These methods sacrifice some accuracy for orders-of-magnitude speed, making them appropriate for exploratory screening, high-throughput computational experiments, and the long-time-scale dynamics of large biomolecules and nanostructures.
Multiscale and hybrid methods
Many real problems span length and time scales that no single method can handle. Hybrid quantum-mechanics/molecular-mechanics (QM/MM) schemes treat a small reactive region, such as an enzyme active site or a defect in a solid, with quantum chemistry while embedding it in a much larger classical environment. Output from electronic-structure calculations also feeds molecular dynamics simulations through ab initio molecular dynamics or, increasingly, through machine-learning interatomic potentials trained on quantum-chemical data.
Comparing Quantum Chemistry Methods
No single method dominates across all problems. The choice is a triangulation between accuracy, computational cost, and the chemistry of the system. The table below summarizes the most widely used families; cost scaling is given as a power of the number of electrons or basis functions, and typical accuracy is approximate and depends on system and property.
| Method family | Cost scaling | Typical accuracy (energies) | Strengths | Common limitations |
|---|---|---|---|---|
| Semiempirical / tight binding | ~ N2–N3 | ~ 5–10 kcal/mol | Very fast, treats thousands of atoms, good for screening and dynamics | Element- and system-dependent parameterizations; poor for unusual bonding |
| Hartree-Fock | ~ N4 | Lacks electron correlation beyond exchange | Conceptual baseline, exact exchange, well-defined wavefunction | Often poor for relative energies; inadequate for thermochemistry and reactions on its own |
| Density functional theory (DFT) | ~ N3 (linear scaling possible) | Often ~ 2–5 kcal/mol for suitable systems and functionals | Best balance of cost and accuracy for many problems; scalable to large molecular and materials models | Can fail outside a functional's domain; self-interaction error, dispersion, strong correlation, charge-transfer states |
| MP2 and post-HF perturbation | ~ N5 | Often ~ 1–3 kcal/mol, system-dependent | Captures basic correlation; useful for non-covalent interactions | Can overbind dispersion and diverge for small-gap or multireference systems |
| Coupled cluster, CCSD(T) | ~ N7 | ~ 1 kcal/mol (chemical accuracy) | Gold-standard accuracy for single-reference closed-shell molecules | Expensive; unreliable for strong static correlation |
| Multireference methods (CASSCF, CASPT2, MRCI) | Combinatorial in active space | Often ~ 1–3 kcal/mol, system-dependent | Handle bond breaking, transition metals, excited states | Active-space selection is expert-driven; cost grows quickly |
| Quantum Monte Carlo | ~ N3–N4 | Can approach benchmark accuracy within fixed-node error | High-quality total-energy benchmarks; parallelizes well; handles strong correlation | Stochastic noise; trial wavefunction quality matters |
| GW / Bethe-Salpeter | ~ N4–N6 | Often ~ 0.1–0.3 eV on bandgaps after careful convergence | Quasiparticle bandgaps and optical spectra of solids and nanostructures | Sensitive to starting point, self-consistency, basis, and convergence settings |
As a rule of thumb, DFT is the default choice for nanoscale materials, surfaces, defects, and screening studies; CCSD(T) is used for high-accuracy molecular benchmarks when the electronic structure is dominated by a single reference; multireference methods are needed for bond breaking, transition-metal complexes, near-degenerate states, and many excited-state problems; GW and Bethe-Salpeter methods are used for quasiparticle bandgaps and optical spectra; and semiempirical, tight-binding, or machine-learning potentials are used when the system size or simulation time is too large for DFT.
In practice, a study often combines methods: high-accuracy coupled cluster or quantum Monte Carlo provides reference values on small models, DFT extends those benchmarks to realistic system sizes, and tight-binding or machine-learning surrogates take care of long simulations and high-throughput screening.
Why Quantum Chemistry Matters at the Nanoscale
Nanoscale objects sit in the regime where the discrete electronic structure of a finite cluster begins to merge with the continuous bands of a bulk solid. Optical color, magnetic ordering, electrical conduction, and chemical activity all depend on which electronic states are filled and how they hybridize across atoms. Quantum confinement in semiconductor nanocrystals, the appearance of spin-polarized edge states in graphene nanoribbons, and the molecule-like discrete levels of an artificial atom all reflect quantum behavior that classical force fields cannot reproduce.
A second reason is that nanoscale systems sit at sizes where quantum chemistry is computationally feasible. A 3 nm quantum dot may contain a few hundred to a few thousand atoms and is often within reach of modern DFT, depending on composition, surface ligands, and the chosen level of theory. A small organometallic catalytic site or a defect in a two-dimensional material may also be small enough for CCSD(T) or quantum Monte Carlo benchmarks. This sweet spot, between molecules small enough for high accuracy and crystals large enough to display bulk-like collective behavior, is where electronic-structure theory is most predictive and where most modern nanochemistry theory operates.
Applications in Nanoscience and Materials
Catalysts and surface chemistry
DFT calculations of binding energies and reaction barriers on metal surfaces and on metal-oxide nanoparticles have become a standard tool in heterogeneous catalysis. Volcano plots and scaling relations between adsorption energies, both pioneered with quantum-chemical data, guide the design of active surfaces and the selection of single-atom catalysts and nanocatalysts. The same methods underlie modern computational design of photocatalysts for water splitting and CO2 reduction, where band-edge alignment, surface adsorption, and excited-state dynamics all need quantum-chemical input.
Quantum dots, optical materials, and bandgap engineering
Time-dependent DFT and GW/Bethe-Salpeter calculations predict the absorption and emission spectra of semiconductor nanocrystals as a function of size, shape, surface ligands, and core/shell architecture. They also rationalize the size-dependent color shifts that define quantum-dot light-emitting diodes. Bandgap engineering in alloys, heterostructures, and doped semiconductors relies on first-principles calculations to forecast how composition, strain, and defects shift the conduction and valence bands.
Two-dimensional materials and defects
DFT and many-body methods explain why graphene is a zero-gap semimetal, why monolayer MoS2 has a direct gap while its bulk form has an indirect gap, and how stacking, twisting, or strain reshape the band structure of 2D materials. Atomic vacancies and other point defects introduce defect states in the bandgap, and rational defect engineering increasingly relies on quantum-chemical input to choose dopants, vacancies, and substitutional sites that give the desired electronic or magnetic behavior.
Energy storage and batteries
DFT-based calculations of voltages, ion migration barriers, and decomposition pathways have reshaped lithium-ion and post-lithium battery research, particularly for solid-state electrolytes and high-voltage cathodes. Computational screening of crystal structures across the Materials Project and similar databases would not be possible without efficient electronic-structure methods.
Quantum sensing, color centers, and quantum technology
The negatively charged nitrogen-vacancy center in diamond and analogous defects in silicon carbide and hexagonal boron nitride are workhorses of solid-state quantum sensing. Predicting their level structure, spin properties, and optical transitions requires beyond-DFT treatment and is one of the most active arenas for high-accuracy quantum chemistry of solids.
Modern Frontiers: Machine Learning and Quantum Computing
Two developments are reshaping quantum chemistry. The first is the integration of machine learning. Neural-network interatomic potentials trained on DFT or coupled-cluster data now reproduce reference forces and energies at a fraction of the cost, enabling molecular-dynamics simulations on nanosecond and microsecond time scales while preserving quantum-chemical accuracy. Machine-learning models also build surrogate property predictors across large materials databases, learn improved exchange-correlation functionals, and provide highly expressive neural-network ansatzes for variational Monte Carlo, with several recent architectures reaching benchmark accuracy on small molecules without the use of a finite basis set.
The second is quantum computing. Because the cost of representing a many-electron wavefunction grows exponentially with the number of electrons on a classical computer but only linearly with the number of qubits on an idealized quantum machine, quantum chemistry is one of the leading proposed applications of quantum hardware. Variational quantum eigensolvers and quantum phase estimation algorithms have been demonstrated on small molecules including hydrogen chains and lithium hydride. Real chemistry beyond the reach of classical methods will require fault-tolerant quantum computers with many millions of physical qubits, and so far quantum-computing chemistry remains an active research field rather than a routine tool.
Limitations and Open Challenges
Despite a century of development, several long-standing problems remain. Strongly correlated electronic systems, such as transition-metal oxides, lanthanide and actinide compounds, and high-temperature superconductors, are poorly described by single-reference methods and stress every approximation in the toolkit. Excited states, especially charge-transfer and Rydberg states, require careful method choice and remain a frontier even for established functionals. Long-range dispersion interactions are not captured by standard local or semi-local DFT and need explicit corrections. Self-interaction error in approximate functionals distorts barrier heights and the description of localized states. Finally, quantum chemistry of complex environments — solvated catalysts, biomolecules, electrochemical interfaces, and ligand-covered nanostructures — demands rigorous coupling between high-accuracy electronic structure, solvent or embedding models, statistical mechanics, and macroscopic transport. The trajectory of the field over the next decade is set by these challenges, and by the increasingly tight integration of quantum chemistry with machine learning, materials databases, and emerging quantum hardware.
FAQ: Quantum Chemistry
What is the difference between quantum chemistry and computational chemistry?
Quantum chemistry is the application of quantum mechanics to chemical problems, whether the work is analytical or numerical. Computational chemistry is the broader practice of using computers to model chemical systems and includes quantum chemistry alongside force-field-based molecular mechanics, classical molecular dynamics, and cheminformatics. In practice the two terms overlap heavily, since almost all modern quantum chemistry is done on computers.
What is the Born-Oppenheimer approximation?
The Born-Oppenheimer approximation separates the motion of electrons from that of nuclei in a molecule. Because nuclei are at least 1836 times heavier than electrons, the electrons relax effectively instantaneously to any nuclear arrangement, so the electronic Schrödinger equation can be solved at fixed nuclear positions. The resulting electronic energies, plotted as a function of nuclear coordinates, define the potential energy surfaces on which chemistry happens. Almost every routine quantum-chemistry calculation invokes this approximation; it breaks down for processes such as nonadiabatic dynamics, ultrafast photochemistry, and conical intersections, which require explicit coupling of electronic and nuclear motion.
Is density functional theory considered an ab initio method?
Strictly speaking, density functional theory is exact in principle through the Hohenberg-Kohn theorems, but every practical calculation uses an approximate exchange-correlation functional whose form is partly empirical. Some authors therefore reserve "ab initio" for wavefunction methods like Hartree-Fock and coupled cluster and refer to DFT separately. Others note that DFT is just as parameter-free as Hartree-Fock at the level of formalism and call it ab initio. The terminology is a matter of convention rather than physics.
What is a basis set in quantum chemistry?
A basis set is the set of mathematical functions used to represent the molecular orbitals or electron density in a calculation. Most molecular quantum chemistry uses Gaussian-type orbitals centered on atoms, ranging from minimal sets such as STO-3G to large correlation-consistent families such as cc-pVQZ. Solid-state codes more often use plane waves combined with pseudopotentials. Larger basis sets give more flexibility and lower energies but increase cost; controlling and reporting the basis set is essential because incomplete basis sets cause systematic errors.
How accurate is density functional theory in practice?
For many ground-state properties of organic molecules and inorganic solids, modern density functional approximations reach errors of a few kilocalories per mole in reaction energies and a few percent in lattice constants and elastic moduli, which is sufficient for many applied screening and interpretation tasks. Accuracy depends on the functional chosen, the property being calculated, and the system. Strongly correlated electrons, weak dispersion interactions, charge-transfer excited states, and reactions involving partially filled d or f shells remain well known weak spots that often require beyond-DFT methods.
What software is used for quantum chemistry calculations?
Many widely used codes implement the standard methods. Molecular work is dominated by packages such as Gaussian, ORCA, Q-Chem, Molpro, NWChem, and PySCF, each with different strengths in wavefunction methods, DFT, and excited states. Solid-state and materials calculations more often use plane-wave codes such as VASP, Quantum ESPRESSO, ABINIT, and CP2K. The right choice depends on system size, basis, and the specific method needed for a given problem.
Further Reading
Physical Review, Self-Consistent Equations Including Exchange and Correlation Effects
The Journal of Chemical Physics, Perspective: Fifty years of density-functional theory in chemical physics
Reviews of Modern Physics, Density functional theory: Its origins, rise to prominence, and future
Reviews of Modern Physics, Quantum computational chemistry
